




Sickle cell retinopathy: diagnosis, management, and treatment

 Save as PDF
Save as PDFABSTRACT
Background
Hemoglobinopathies, such as sickle cell disease, are characterized by abnormal or insufficient production of hemoglobin chains that lead to complications such as anemia, stroke, asthma, osteoporosis, infection, and vision loss. This case report will discuss the epidemiology, diagnostic testing, management, and treatment options for sickle cell retinopathy.
Case report
A 45-year-old African American male presented for a routine eye exam with a chief complaint of intermittent floaters OU. His systemic history was significant for sickle cell disease with a history of chronic anemia managed with folic acid and vitamin D supplementation. Fundus examination and diagnostic testing with OCT revealed proliferative retinopathy due to sickle cell-hemoglobin C disease.
Conclusion
Sea fan neovascularization is a well-described phenomenon associated with proliferative sickle cell retinopathy. Sea fan and peripheral neovascularization have also been implicated in other proliferative retinopathies, such as sarcoidosis and Eales disease. Proliferative retinopathy is a sight-threatening condition which requires diagnostic differentiation to provide proper treatment for ocular and systemic findings.
Keywords: Sickle cell retinopathy, sickle cell disease, neovascularization, hemoglobin (Hb)
INTRODUCTION
Sickle cell disease is an inherited group of blood disorders that affect the hemoglobin in red blood cells, a protein that helps carry oxygen throughout the body. Normally, red blood cells are disc-shaped and flexible in order to move through blood vessels. However, in those with sickle cell disease, the red blood cells are stiffened, crescent or sickle-shaped, and unable to bend or navigate through blood vessels. As a result of this structural change, functional damage to organs occurs due to vaso-occlusion, hemolysis, and tissue ischemia.1 The ophthalmic manifestations of sickle cell retinopathy fall into two categories:
- Non-proliferative sickle cell retinopathy
- Vascular tortuosity
- Pigmented lesions with spiculated borders (commonly known as black sunburst)
- Pink intraretinal hemorrhage (commonly known as salmon-patch)
- Angioid streaks
- Schisis cavity with refractile elements (iridescent spots)
- Proliferative sickle cell retinopathy
- Stage I: Peripheral arteriolar occlusions
- Stage II: Peripheral arteriolar-venular anastomoses
- Stage III: Neovascular and fibrous proliferations (commonly known as sea fan)
- Stage IV: Vitreous hemorrhage
- Stage V: Tractional retinal detachment
The most common cause of blindness in people with sickle cell disease is proliferative sickle cell retinopathy.2 Progression is typically associated with age, extent of sickle cell retinopathy, and/or presence of retinopathy in the contralateral eye.2 Therefore, co-management with specialists such as hematologists and retinal specialists is important to reduce the risk of additional systemic complications and to provide appropriate ocular treatment including procedures such as injections, laser, and/or surgery.
CASE REPORT
A 45-year-old African American male presented for an eye exam with complaints of intermittent floaters and blurred vision in both eyes. Upon questioning, the floaters had been present for the last three years. He denied photopsia, eye pain or photophobia.
His past medical history included bilateral hearing loss and tinnitus, major depressive disorder, vitamin D deficiency and sickle cell disease. He was taking: folic acid, vitamin D, and a multivitamin without iron. He was first diagnosed with sickle cell disease at birth in 1977 and had a single episode of sickle cell crisis requiring hospitalization at the age of 14. He denied any history of previous blood transfusions.
He was evaluated in the hematology clinic about 5 years prior and was confirmed to have sickle cell disease with a positive sickle solubility test and heterozygosity for HbS and HbC via high performance liquid chromatography (HPLC) and hemoglobin electrophoresis. Results of his testing are listed below in Table 1.

Table 1. Lab results for sickle cell disease. Confirms diagnosis of hemoglobin SC disease.
Regarding his family history, his father died at age 51 from a heart attack, while his mother died at 59 from a stroke. Both of his parents were reported to have sickle cell trait. He had four children, 3 of whom are male and 1 female. His oldest child has no history of sickle cell disease, the second-oldest has sickle cell disease, and the younger two have sickle cell trait. One of his four sisters had a “more severe form” of sickle cell disease with a history of multiple hospitalizations for vaso-occlusive crises.
On review of systems, he denied any current fever, chills, cough, blood in urine, unusual weight loss or gain, trouble breathing or chest pain. Upon further questioning, he reported hip and back pain. He denied any recent trauma or surgery. Specifically, he denied any history of splenectomy or cholecystectomy.
On initial examination, his best corrected visual acuity (BCVA) was 20/20- in the right eye and 20/20 in the left eye. Pupil examination, confrontation visual fields and extraocular movements were normal in both eyes. Slit lamp examination was also normal in both eyes. Intraocular pressures were 22mmHg in the right eye and 24mmHg in the left eye via non-contact tonometry.
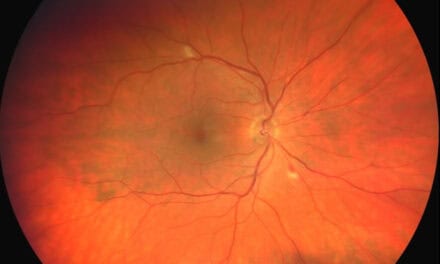
The fundus exam for the right eye was normal, but the fundus exam for the left eye showed pigment mottling of the macula and a pre-retinal fibrovascular lesion in the temporal periphery. Additional fundus images are shown below in Figures 1 and 2.

Figure 1: Color fundus photos: The right eye appears grossly normal.
Figure 2: Color fundus photos (A, B): (A) Pigment mottling of the retina in the left eye. (B) Pre-retinal fibrovascular lesion in the temporal periphery
Given the fundus findings with his medical and family history, the primary diagnosis was proliferative sickle cell retinopathy OS. Other differential diagnoses considered were ocular ischemic syndrome, Eales disease, proliferative diabetic retinopathy, sarcoidosis, vein occlusion, and embolic retinopathy. At this time, we recommended that he continue his care with hematology to lower the risk of systemic complications. He was also referred to a retinal specialist for further evaluation and treatment of neovascularization.
Follow-up #1 with Ophthalmology
The patient returned one month later for his follow-up with the retinal specialist to evaluate and determine treatment for sickle cell retinopathy. He denied any new visual complaints. His visual acuity without correction was 20/30, PH to 20/20 OD, 20/25-2 OS. His eye pressure was within normal limits, and his slit lamp exam was unremarkable for any new anterior segment findings. Upon dilated fundus examination, his right eye was unremarkable for any new retinal findings and his optic nerve was healthy with distinct margins. However, his left eye showed a small and stable retinal pigment epithelial detachment near the fovea. Additionally, his peripheral retinal exam showed a pre-retinal fibrotic lesion that was consistent with a regressed neovascular frond in the temporal arcade and associated with an intraretinal hemorrhage. The diagnosis of proliferative sickle cell retinopathy OS was confirmed, and he was scheduled to return for photos, OCT, and fluorescein angiography.
Follow-up #2 with Ophthalmology
The patient returned two months later for his follow-up with ophthalmology to evaluate his sickle cell retinopathy. He reported no changes to vision. He denied any new flashes or floaters. His visual acuity without correction was 20/40, PH to 20/20-1 OD, 20/25 OS. His eye pressure was within normal limits, and his slit lamp exam was unremarkable for any new anterior segment findings. Dilated fundus examination was normal in the right eye (Fig. 3A). His left eye showed a stable neovascular frond in the temporal arcade (Fig. 3B). On the same day, fluorescein angiography was performed, which confirmed the presence of a proliferative complex with a surrounding area of capillary non-perfusion in the left eye. The right eye was also found to have patchy areas of capillary non-perfusion in the temporal retina (Fig. 4A-B). At this time, treatment was recommended for the left eye with panretinal photocoagulation.

Figure 3: (A) Color fundus photo of right eye. (B) color fundus photo showing stable area of neovascularization in the temporal arcade consistent with proliferative sickle cell retinopathy.

Figure 4 (A,B).Late phase fluorescein angiograms illustrate areas of patchy capillary non-perfusion in the temporal peripheral retina.
Follow-up #3 with Ophthalmology
The patient reported no changes to his vision at this visit and denied flashes, floaters, or any eye pain. His visual acuity without correction was 20/30 OD, 20/25 OS. His eye pressure was within normal limits, and he was dilated with tropicamide and phenylephrine. Scattered panretinal photocoagulation was applied to his left eye using a 200-micron sized spot laser at a power of 100-110mW to apply 265 spots at the site of neovascularization in the temporal periphery. The patient tolerated the procedure well and post-treatment imaging was performed as depicted in Figure 6. He was advised to follow-up in two months for repeat dilation and evaluation for additional treatment if necessary.

Figure 6. Panretinal photocoagulation applied to the left eye in a sectoral pattern around the fibrotic neovascular frond.
DISCUSSION
Sickle cell disease is an autosomal recessive genetic disorder that was discovered in 1910 and continues to be a major public health concern. Hemoglobinopathies are among the most common genetic disorders worldwide and are transmitted as autosomal recessive disorders from parents who carry abnormal hemoglobin genes. When an individual inherits a hemoglobin A gene from each parent to form HbAA, this is considered a normal inheritance pattern. Sickle cell disease is caused by inheriting hemoglobin chains that have a single point mutation in the hemoglobin beta subunit. Specifically, a single mutation at the sixth position of the β-globin chain induces a substitution of valine for glutamic acid, resulting in hemoglobin S, an abnormal allele of hemoglobin.3 These unique genotypic inheritance patterns dictate the phenotypic variations and ultimately the clinical presentation of systemic complications. As a result, genetic counseling is a crucial factor in determining effective therapy and proper management of complications associated with sickle cell disease.
The most common abnormal genotypes of sickle cell hemoglobinopathies (Hb) include:
- AS (sickle cell trait)
- SS (sickle cell anemia)
- CC (hemoglobin C disease)
- SC (sickle cell-hemoglobin C disease)
- AC (hemoglobin C trait)
- Beta thalassemia: hemoglobin S (HbS) or S-beta thalassemia (SThal) disease
Due to its natural protection against malaria, the sickle cell trait is more common in people of African heritage or with ancestors from tropical/subtropical areas.4 Approximately one in every thirteen African American children born in the United States has the sickle cell trait.5 In sickle cell trait, the hemoglobin A:S ratio is usually approximately 60:40. In episodes of ischemia, red blood cells containing HbS will undergo reversible changes in their cell membrane. This alters the morphology of the cell and changes it into a sickle shape, increasing the risk of vaso-occlusion and focal ischemia. Most patients with sickle cell trait are asymptomatic, as their volume of RBCs with HbS is low. They do not have a vaso-occlusive crisis unless subjected to severe oxidative stress, such as severe hypoxia, dehydration, elevated sympathetic outflow, and/or release of inflammatory cells.4
HbSS, HbSC, and Hb SThal will be our primary focus for discussion among the genotypes indicated above because they have a higher prevalence. It is estimated that one in every 365 African Americans in the United States has sickle cell disease (SCD).5 The likelihood of African Americans developing sickle cell disease and presenting with HbSS, HbSC, or HbSThal is 0.2%, 2% and 0.03%, respectively.6 HbSS is the most severe and prevalent form of SCD, accounting for 65% of cases, while HbSC accounts for an estimated 30% of cases.6 Acute chest syndrome (ACS) is the most common complication and cause of death in SCD patients, particularly those with HbSS and HbSThal. This condition is triggered by an acute vaso-occlusive event and patients may present with chest pain, a sudden cough and/or shortness of breath that can lead to respiratory failure if not treated as soon as possible. Other common complications of sickle cell disease include general fatigue, muscle weakness, increased risk for infections, leg ulcers, and/or stroke. These systemic complications can vary in severity depending on the genotype, and can become debilitating or even life threatening if not managed appropriately.
Sickle cell anemia typically has more severe systemic complications, while HbSC is more likely to develop severe ocular complications. It’s hypothesized that the reason for this paradox is that the enhanced circulatory competence of HbSC cells preserve the retinal circulation and allow for the development of proliferative lesions if retinal ischemia were to occur.7 On the other hand, those who have sickle cell anemia will have an earlier and more complete occlusion of the peripheral retinal vessels so smaller vessel damage and proliferative lesions are less likely.7 In general, sickle cell retinopathy can develop in up to 42% of those with SCD during their second decade of life, and it affects an estimated 15-20% of those with HbSS and 33-40% of those with HbSC in the United States.6 Patients with HbSC, HbSS, or HbSThal in the United States are 33%, 3%, and 14% more likely to develop proliferative sickle cell retinopathy, respectively.6
Most patients who have sickle cell disease are asymptomatic for visual changes, mostly since the macula is rarely affected. However, those who are symptomatic may experience a decrease in vision, loss of visual field, floaters, flashes, scotomas, or changes to color vision. A hallmark finding of sickle cell disease is sea-fan neovascularization that presents as a frond-like configuration that favors growth in the periphery of the superior-temporal peripheral retina, especially superior temporal. Sea-fan neovascularization occurs around 18 months following the development of arterio-venular anastomosis and it may even become fibrotic and regress on its own in up to 60% of cases due to auto-infarction.8 This spontaneous regression, however, may not occur until around two years following the onset of proliferative sickle cell retinopathy.7 Persistent neovascularization can result in bands of vitreous tension and the development of vitreous hemorrhage, which affects 21-23% of people with HbSC.8 If the vitreous traction continues to contract, it may result in additional retinal complications such as a tractional retinal detachment.
In the United States, screening for sickle cell disease in newborns is mandated in all 50 states and the District of Columbia since 2008.9 An estimated 4000 to 5000 pregnancies are at risk for sickle cell disease and patients can be screened as early as birth.10 The solubility sickling tests is a relatively inexpensive test that is used to detect the polymerization of HbS in a deoxygenated state, but it is unable to differentiate between sickle cell trait and sickle cell disease.11 Hemoglobin electrophoresis is utilized to differentiate and detect sickle cell disease by using three different tests to detect Hb variants and differentiate between HbS and HbC.11 Isoelectric focusing (IEF) is considered the standard test for newborn screening since it only needs a small sample. It is a method for separating proteins depending on their isoelectric points which helps to detect HbS and HbA even in the presence of high concentrations of HbF which is high in newborns but not in adults.11 Lastly, high performance liquid chromatography (HPLC) is a highly sensitive test that can be fully automated and reliable even when patients are undergoing blood transfusions. It is the preferred method of diagnosis and able to detect hemoglobin variants such as HbF, Hb A2, HbS, HbC, HbBarts based on retention time and shape of peak.11
For many years, the main preventative treatment has been hydroxyurea to help reduce the likelihood of complications from sickle cell disease, including sickle cell retinopathy.12 Hydroxyurea reduces rigidity in red blood cells by increasing fetal hemoglobin F production inhibiting hemoglobin polymerization. Blood transfusions are also used in both acute and chronic systemic care to increase the circulation of normal red blood cells and reduce the amount of sickle hemoglobin red blood cells which helps to reduce risk of stroke. Other newer oral medication options include voxelotor and L-glutamine powder which may be used in adults and children to prevent red blood cells from turning into a sickle shape and neutralize oxidative stress which helps keep red blood cells in a flexible state and improve blood flow.13 However, the only cure for sickle cell disease is a blood and bone marrow transplant. It is typically reserved for severe cases in children, and has shown to have a success rate as high as 80% when the donor is related to the recipient and with matched human leukocyte antigen (HLA).14
Patients with sickle cell disease should be screened as early as 10 years old for ocular complications, especially those with HbSC, HbSS, and HbSThal, to identify any retinal lesion, visual loss, and presence of non-proliferative and/or proliferative sickle cell retinopathy.15 If patients are asymptomatic with no ocular findings, they may be monitored in one-to-two-year intervals.16 However, in cases of proliferative sickle cell retinopathy, these patients should be referred to a retinal specialist. Currently, scatter and feeder vessel photocoagulation are the recommended treatment options for sea fan neovascularization. Scatter laser photocoagulation can be sectoral or circumferential, with treatment administered at the site of neovascularization or 360 degrees to the entire peripheral retina. Feeder vessel photocoagulation involves the application of direct and highly concentrated laser burns to the feeding arterioles, resulting in the closure of neovascular fronds. Although both approaches are effective, scatter laser photocoagulation has been demonstrated to be both safer and more effective than focal laser photocoagulation in decreasing and causing neovascularization regression.17 If neovascularization is persistent, anti-VEGF may be administered in conjunction with laser. When a retinal detachment occurs, surgical options such as a pars plana vitrectomy and/or scleral buckle are recommended.
CONCLUSION
Sickle cell disease is one of the world’s most common hemoglobinopathies, with a wide spectrum of systemic manifestations.6 The most common ocular manifestations of sickle cell disease are non-proliferative and proliferative sickle cell retinopathy. Approximately 5-20% of people with proliferative sickle cell retinopathy will have permanent vision loss.3 The hallmark sign of proliferative retinopathy is sea fan neovascularization caused by retinal ischemia. Depending on the extent of proliferative retinopathy, sickle cell retinopathy may require treatments such as anti-VEGF, laser photocoagulation, or surgical surgery, which requires timely co-management with ophthalmology. Eye care providers must also coordinate with hematology to determine an individual’s genotype, if unknown, using lab testing such as hemoglobin electrophoresis and HPLC to better understand the risk of progression for both ocular and systemic complications. This can aid in the development of a systemic management plan with other specialties, such as cardiology or pulmonology, who design treatment strategies for both acute and chronic care, such as oral drugs or blood transfusions.
REFERENCES
- Williams TN, Thein SL. Sickle Cell Anemia and Its Phenotypes. Annu Rev Genomics Hum Genet. 2018 Aug 31;19:113-147.
- Downes SM, Hambleton IR, Chuang EL, Lois N, Serjeant GR, Bird AC. Incidence and natural history of proliferative sickle cell retinopathy: observations from a cohort study. Ophthalmology. 2005 Nov;112(11):1869-75.
- Abdalla Elsayed, M.E.A., Mura, M., Al Dhibi, H. et al. Sickle cell retinopathy. A focused review. Graefes Arch Clin Exp Ophthalmol 257, 1353–1364 (2019).
- Ashorobi D, Ramsey A, Yarrarapu SNS, et al. Sickle Cell Trait. [Updated 2022 Jul 18]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2023 Jan.
- Mangla A, Ehsan M, Agarwal N, et al. Sickle Cell Anemia. [Updated 2022 Nov 30]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2023 Jan.
- Menaa F, Khan BA, Uzair B, Menaa A. Sickle cell retinopathy: improving care with a multidisciplinary approach. J Multidiscip Healthc. 2017 Aug 30;10:335-346.
- Mônica Barbosa de Melo, An eye on sickle cell retinopathy, Revista Brasileira de Hematologia e Hemoterapia, Volume 36, Issue 5, 2014, Pages 319-321,
- Friedman, Neil J., author. The Massachusetts Eye and Ear Infirmary Illustrated Manual of Ophthalmology. St. Louis, Missouri :Elsevier, 2021.
- US Preventive Services Task Force. Screening for sickle cell disease in newborns: recommendation statement. Am Fam Physician. 2008 May 1;77(9):1300-2.
- Motulsky AG. Frequency of sickling disorders in U.S. blacks. N Engl J Med. 1973 Jan 4;288(1):31-3.
- Arishi WA, Alhadrami HA, Zourob M. Techniques for the Detection of Sickle Cell Disease: A Review. Micromachines (Basel). 2021 May 5;12(5):519.
- Yawn BP, Buchanan GR, Afenyi-Annan AN, et al. Management of sickle cell disease: Summary of the 2014 evidence-based report by expert panel members. JAMA 2014;312:1033-1048.
- Migotsky M, Beestrum M, Badawy SM. Recent Advances in Sickle-Cell Disease Therapies: A Review of Voxelotor, Crizanlizumab, and L-glutamine. Pharmacy (Basel). 2022 Sep 26;10(5):123.
- Shenoy S. Hematopoietic stem-cell transplantation for sickle cell disease: current evidence and opinions. Ther Adv Hematol. 2013 Oct;4(5):335-44.
- Li J, Bender L, Shaffer J, et al. Prevalence and onset of pediatric sickle cell retinopathy. Ophthalmology 2019;126:1000-1006.
- Kavanagh PL, Fasipe TA, Wun T. Sickle Cell Disease: A review. JAMA. 2022;328(1):57–68.
- Myint KT, Sahoo S, Thein AW, Moe S, Ni H. Laser therapy for retinopathy in sickle cell disease. Cochrane Database Syst Rev. 2015 Oct 9;2015(10):CD010790

Dr. Njeru is a staff optometrist at the Chillicothe VA Medical Center. He graduated from The Ohio State University: College of Optometry and completed his ocular disease residency at the Chillicothe and Columbus VA. . He is a fellow of the American Academy of Optometry.

Dr. Spotts graduated from Pennsylvania College of Optometry in 2001 and went on to complete a primary care residency at Northeastern State University Oklahoma College of Optometry from 2005-2006. He is currently an optometrist at VA Salisbury.

Melissa Chen, OD is currently a staff optometrist at the Charlotte VA Health Care Center. She received her Doctor of Optometry from Salus University, Pennsylvania College of Optometry in 2019. She then went on to complete her residency at the W.G. (Bill) Hefner VA Medical Center. She also serves as an examiner at the National Board of Examiners in Optometry.

{kind=link}