




Case Report: Genetically Confirmed Best Vitelliform Macular Dystrophy and Butterfly Pattern Dystrophy in the Same Family
 Save as PDF
Save as PDFABSTRACT
BACKGROUND
Best vitelliform macular dystrophy (BVMD) is a relatively common inherited retinal disease caused by mutations in the BEST1 gene. Butterfly pattern dystrophy (BPD) is a relatively uncommon form of pattern dystrophy with no known association to BVMD. Recently, mutations in the CTNNA1 gene have been implicated in cases of BPD. Both conditions are inherited in an autosomal dominant manner; however, these diseases exhibit very different phenotypes. To date, no reports exist of concomitant BEST1 and CTNNA1 mutations within the same pedigree, with separate BVMD and BPD phenotypes among family members.
CASE REPORT
A 57-year-old man with non-specific bilateral macular pigmentary changes presented for retinal evaluation. Multimodal retinal imaging, electrodiagnostic testing and genetic sequencing revealed a diagnosis of BPD while harboring pathogenic mutations in both BEST1 and CTNNA1. The patient’s daughter, who previously presented with active bilateral choroidal neovascularization and macular hemorrhaging secondary to BVMD, was also found to harbor the same pathogenic BEST1 and CTNNA1 mutations. Despite identical contributory genetics, each patient exhibited markedly disparate retinal findings with drastically different outcomes.
CONCLUSION
This case report describes, to these writers’ knowledge, the first genetically confirmed case of simultaneous BEST1 and CTNNA1 mutations causing distinct BVMD and BPD phenotypes between family members within the same pedigree. Clinicians should be aware of genotype-phenotype heterogeneity secondary to incomplete penetrance and expressivity in autosomal dominant inherited retinal disease and the risk of choroidal neovascularization in BPD.
Keywords: Best vitelliform macular dystrophy, BEST1, Butterfly pattern dystrophy, CTNNA1
INTRODUCTION
Best vitelliform macular dystrophy (BVMD) is a relatively common inherited retinal disease caused by one of hundreds of known mutations in the BEST1 gene.1,2 BEST1 encodes the protein bestrophin-1.1,2 Dysfunctional bestrophin-1 causes excess accumulation of subretinal fluid and lipofuscin in the macula.1,2 Ultimately, this leads to the development of a sub-foveal vitelliform lesion, which may be appreciated as early as the first decade of life.1-3 Over time, these lesions eventually collapse, leading to complete outer retinal atrophy.1-3 Choroidal neovascularization with subsequent exudation and hemorrhaging is a well-recognized complication of this disease process and is a leading contributor to vision loss in these patients.1-3 Electro-oculogram (EOG) recordings in BVMD classically exhibit a reduced Arden ratio (light peak-to-dark trough ratio), making it an important diagnostic test.1-3
Butterfly pattern dystrophy (BPD), a unique entity to BVMD, is one of five recognized pattern dystrophies of the retinal pigment epithelium (RPE), all of which affect the macula to some extent.4,5 Pattern dystrophies are classically inherited in an autosomal dominant manner through mutations in the PRPH2 gene.4,5,6 In the majority of cases, patients retain excellent vision without complication throughout life, however, reports of vision loss secondary to RPE atrophy or choroidal neovascularization exist.4,6 Thus, visual function across the pattern dystrophies is often highly variable. Recently, mutations in the gene CTNNA1, which encodes α-catenin 1, a protein involved in forming inter-epithelial cell structural adherens junctions, have been recognized as a novel cause of BPD specifically.7
To date, there are no known reports of genetically confirmed simultaneous BEST1 and CTNNA1 pathogenic mutations in multiple individuals within the same family pedigree exhibiting disparate phenotypes classic for either BVMD or BPD. Herein, we report the first such genetically confirmed cases in the same family.
CASE REPORT
A 57-year-old South Asian male, an established patient, presented for follow-up retinal evaluation without any complaints of decreased vision. The patient was previously examined in the clinic six months prior and he reported that there were no changes to his vision since then.
His personal ocular history was remarkable for longstanding bilateral “macular RPE dropout” without a definitive diagnosis and pterygia in both eyes; he was status-post pterygium excision surgery of the right eye five years prior. His personal medical history was remarkable for essential hypertension, hyperlipidemia, pre-diabetes mellitus and hypothyroidism. His medication list consisted of losartan, atorvastatin, metformin and levothyroxine.
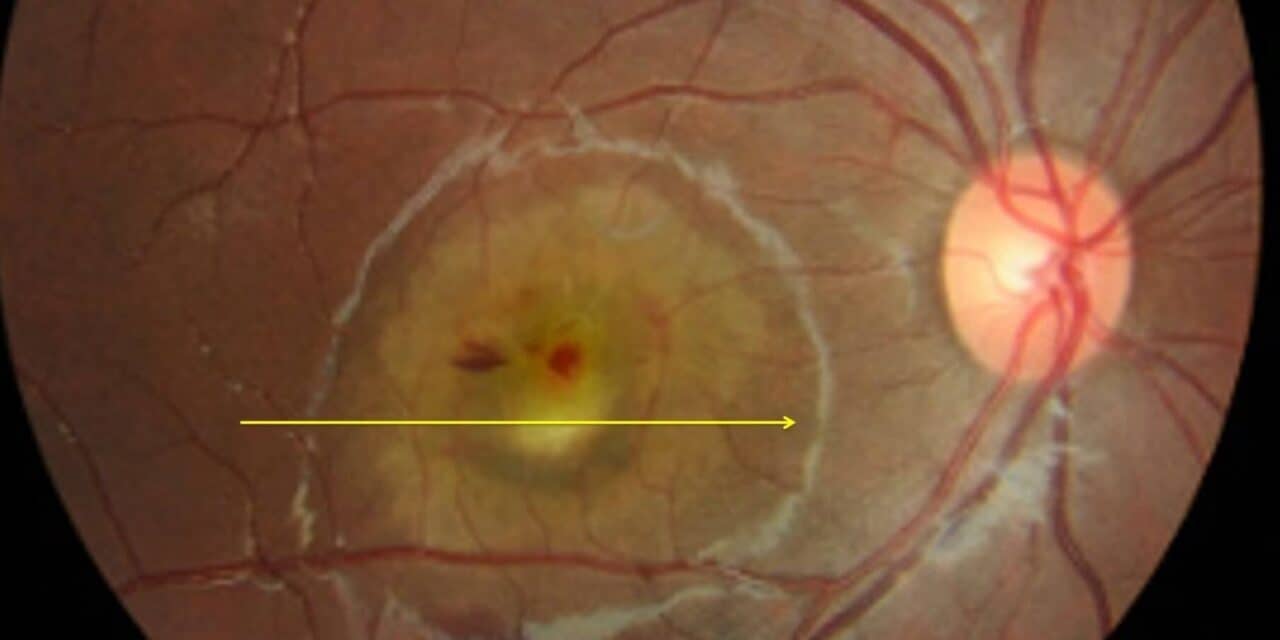
The patient’s family ocular history was noteworthy. First, his daughter was previously diagnosed 15 years prior with an aggressive form of BVMD after developing significant vision loss secondary to bilateral choroidal neovascularization with bilateral subretinal hemorrhaging at six years of age (Figure 1). She eventually underwent numerous rounds of intravitreal anti-vascular endothelial growth factor (anti-VEGF) injections with full resolution and restoration of vision in both eyes. Subsequent genetic testing revealed pathogenic mutations in the BEST1 (c.288G>C (p.Gln96His)) and CTNNA1 (c.965C>T (p.Ser322Leu)) genes.
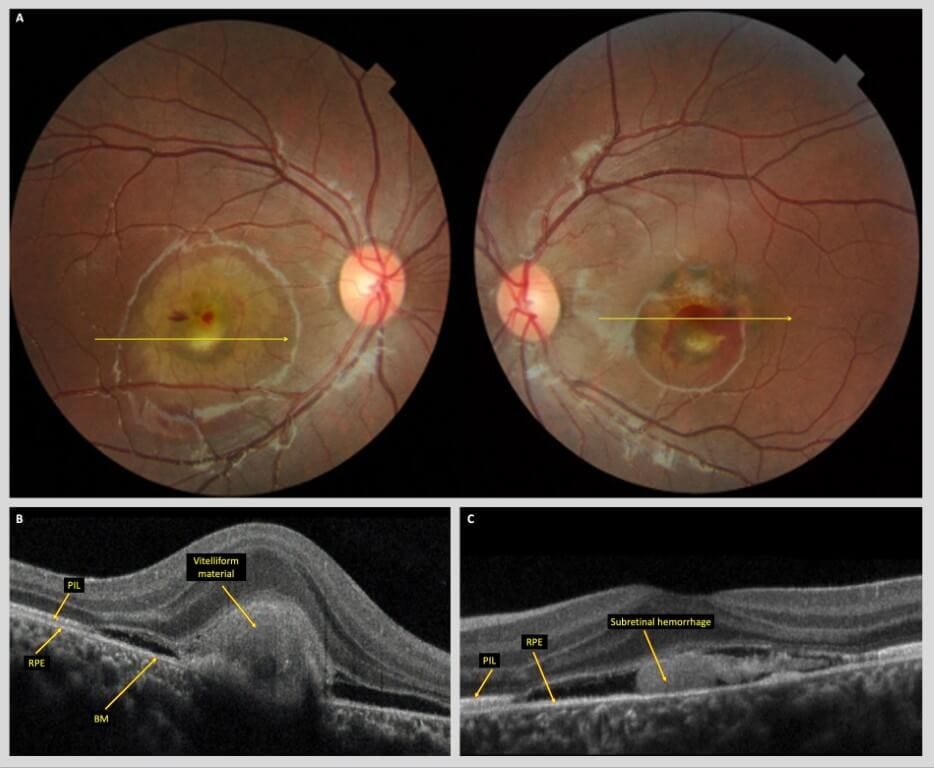
Figure 1. A) Color fundus photos of the right and left eyes of the patient’s daughter upon initial presentation at 6 years of age, demonstrating bilateral subfoveal vitelliform lesions with associated CNVM and subretinal hemorrhaging. Yellow arrows denote location of corresponding raster B scan in panels B and C. B) Spectral-domain optical coherence tomography (SD-OCT) B- scan of the right macula reveals a large subretinal vitelliform lesion with adjacent subretinal fluid. C) SD-OCT B-scan of the left macula reveals subretinal hemorrhage with subretinal fluid and neurosensory detachment. Abbreviations: PIL, photoreceptor integrity line; RPE, retinal pigment epithelium; BM, Bruch’s membrane.
His son, who also underwent genetic and electrophysiologic testing two years prior at the age of 12 following his sister’s diagnosis but who did not develop BVMD, was found to have the same pathogenic CTNNA1 mutation (c.965C>T (p.Ser322Leu)). Notably, the son’s EOG revealed borderline subnormal responses in both eyes (not pictured here) despite the absence of any fundus abnormalities on both clinical examination and multimodal retinal imaging (Figure 2) nor any BEST1 mutations. His visual acuity remained excellent at 20/20 in each eye.
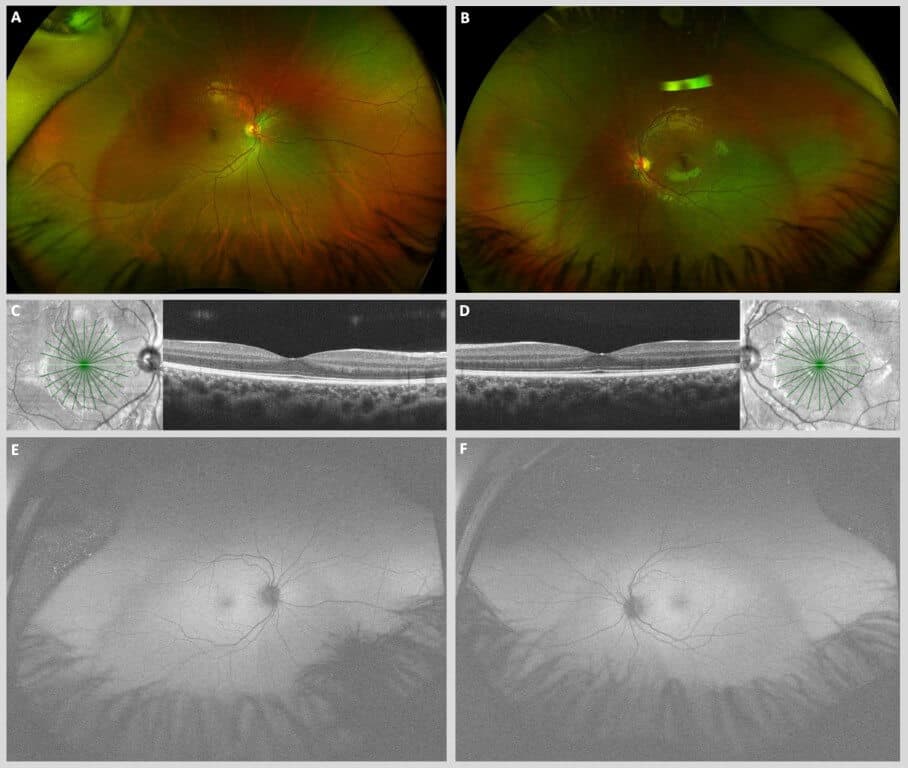
Figure 2. Ultrawidefield pseudo-color fundus photographs of the right (A) and left (B) eyes of the patient’s son demonstrating unremarkable posterior segments in each eye. Macular spectral domain optical coherence tomography (SD-OCT) B-scans of the right (C) and left (D) eyes demonstrate entirely unremarkable maculae with intact foveal contour and retinal laminations. Ultrawidefield fundus autofluorescence (FAF) photographs of the right (E) and left (F) eyes demonstrate normal iso-autofluorescence throughout the posterior segments of each eye.
When we examined the patient described in this case, his uncorrected visual acuities were 20/20-1 in the right and left eyes. Pupils appeared isocoric, round and briskly reactive to light without relative afferent pupillary defect in either eye. Extraocular motility was full in all positions of gaze in both eyes, and gross confrontation fields were full in both eyes. Intraocular pressure as measured with Goldmann applanation tonometry was 18 mmHg and 17 mmHg in the right and left eyes, respectively. Anterior segment slit lamp examination was remarkable for nasal corneal scarring status-post pterygium excision in the right eye, a small pterygium on the left cornea and 1+ nuclear sclerosis of the lens in both eyes.
Dilated fundus examination revealed relatively symmetric yellow linear branching pigmentary lesions in the macula of both eyes (Figure 3A-B). Peripheral retinal examination revealed multiple symmetric large lobulated coalescent areas of RPE hypopigmentation, most prominent in the temporal and inferotemporal far peripheral retina. These lesions were more apparent with fundus autofluorescence (FAF) imaging, which also revealed diffuse and symmetric macular, mid-peripheral and peripheral stippled hyper- and hypo-autofluorescence throughout the posterior segment (Figure 3C-D). High definition spectral-domain optical coherence tomography (SD-OCT) raster scans of the macula revealed bilateral blunted foveal contours in the inner retina, with a notable thickened, undulating, irregular RPE layer throughout the macula with visible underlying Bruch’s membrane and choroid hypertransmission in both eyes (Figure 3E-F). There was a notable absence of classic vitelliform lesions or subretinal fluid in either eye.
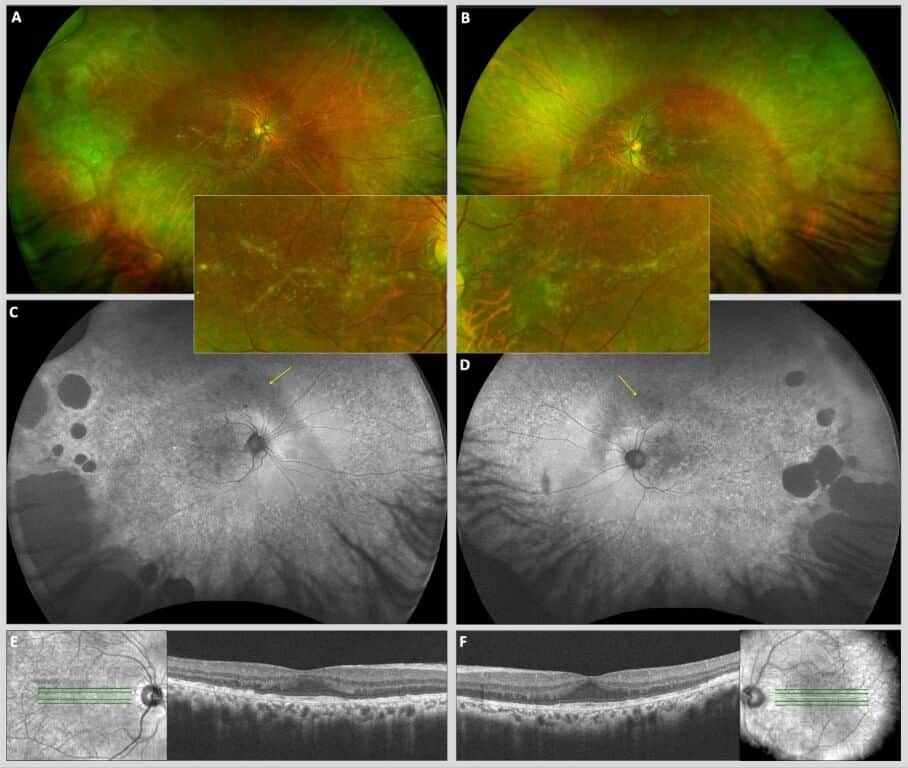
Figure 3. Ultrawidefield pseudo-color fundus photographs of the right (A) and left (B) eyes demonstrating bilateral and symmetric peripheral chorioretinal atrophy and linear, branching, radial macular pigmentary changes consistent with butterfly pattern dystrophy (maculae enlarged in insets). Ultrawidefield fundus autofluorescence (FAF) photographs of the right (C) and left (D) eyes showing diffuse, stippled hyper- and hypo-autofluorescence throughout the entire fundus from posterior pole to far retinal periphery with relatively symmetric large lobulated coalesced areas of hypo-AF peripherally. Symmetric arcuate bands of hypo-autofluorescence are observed superior to the optic nerves (yellow arrows) corresponding to the inferior arcuate visual field defects seen in Figure 4. A hyper-AF signal represents lipofuscin accumulation secondary to dysfunctional outer retina (photoreceptors) and RPE, while a hypo-AF signal represents areas of outer retinal and RPE degeneration and atrophy. Macular spectral domain optical coherence tomography (SD-OCT) B scans of the right (E) and left (F) eyes demonstrating thickened, irregular retinal pigment epithelium (RPE) with visible underlying Bruch’s membrane throughout the macula with blunted foveal contours in each eye. Diffuse choroidal hypertransmission throughout the macula of each eye is also appreciated.
Subsequent standard automated perimetry (Figure 4) demonstrated relatively congruous inferior arcuate scotomata, most dense inferotemporal in both eyes, corresponding to areas of hypo-autofluorescence superonasal to the optic nerve head in both eyes (Figure 3C-D).
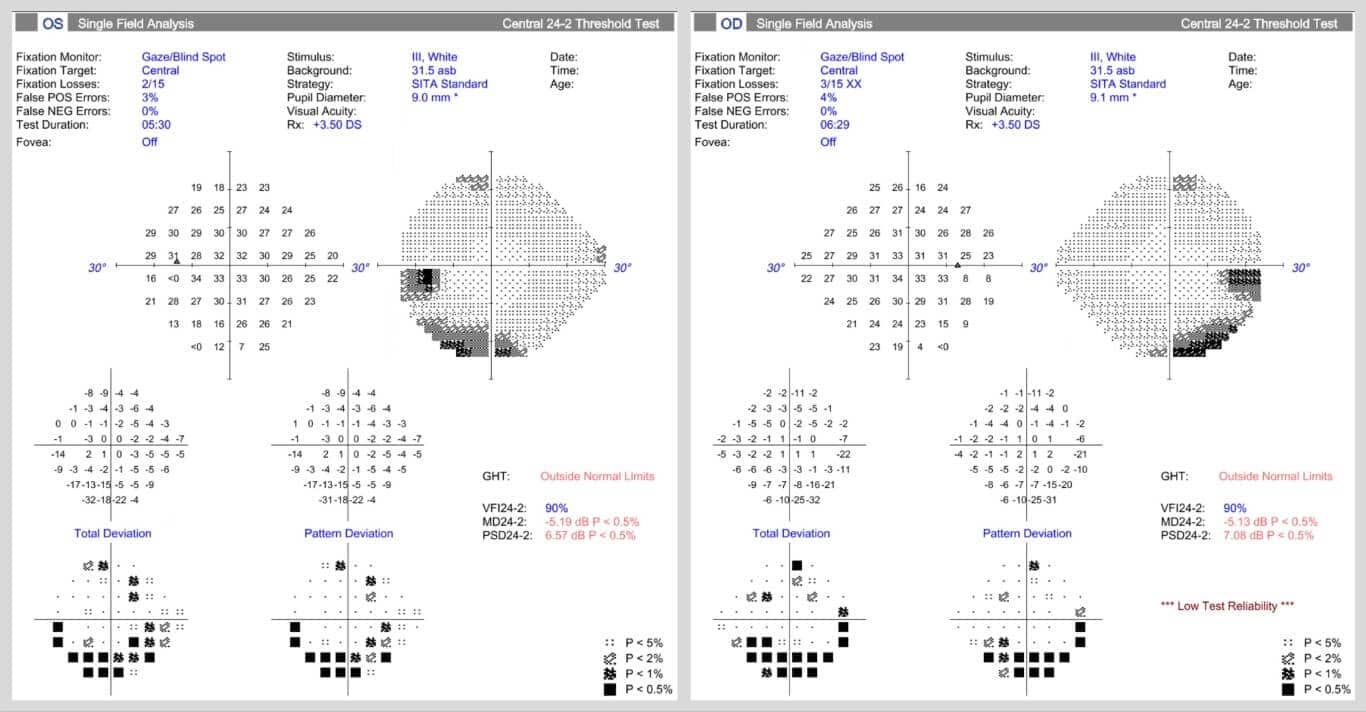
Figure 4. Standard automated perimetry Humphrey visual field 24-2 paradigm utilizing the SITA standard thresholding algorithm of the right and left eyes, demonstrating symmetric inferior arcuate scotomata corresponding to areas of superior hypo-autofluorescence on ultra-widefield fundus autofluorescence images, as in Figure 3C & D.
Electrophysiologic testing was performed, including full-field flash electroretinography (ERG) and EOG. For full-field ERG, light-adapted photopic stimuli were first presented, followed by thirty minutes of dark adaptation before dark-adapted scotopic stimuli were subsequently presented. Otherwise, all other aspects of electrophysiologic testing were conducted in accordance with the International Society for Clinical Electrophysiology of Vision (ISCEV) standard protocol following pupillary mydriasis.8
ERG testing illustrated globally attenuated but not extinguished photopic and scotopic responses in both eyes (Figure 5). EOG testing was significant for a markedly reduced light peak-to-dark trough (Arden) ratio of 1.10 and 1.11 in the right and left eyes, respectively (Figure 6; normal Arden ratio: >1.80; subnormal: 1.60 – 1.80; reduced: < 1.60).
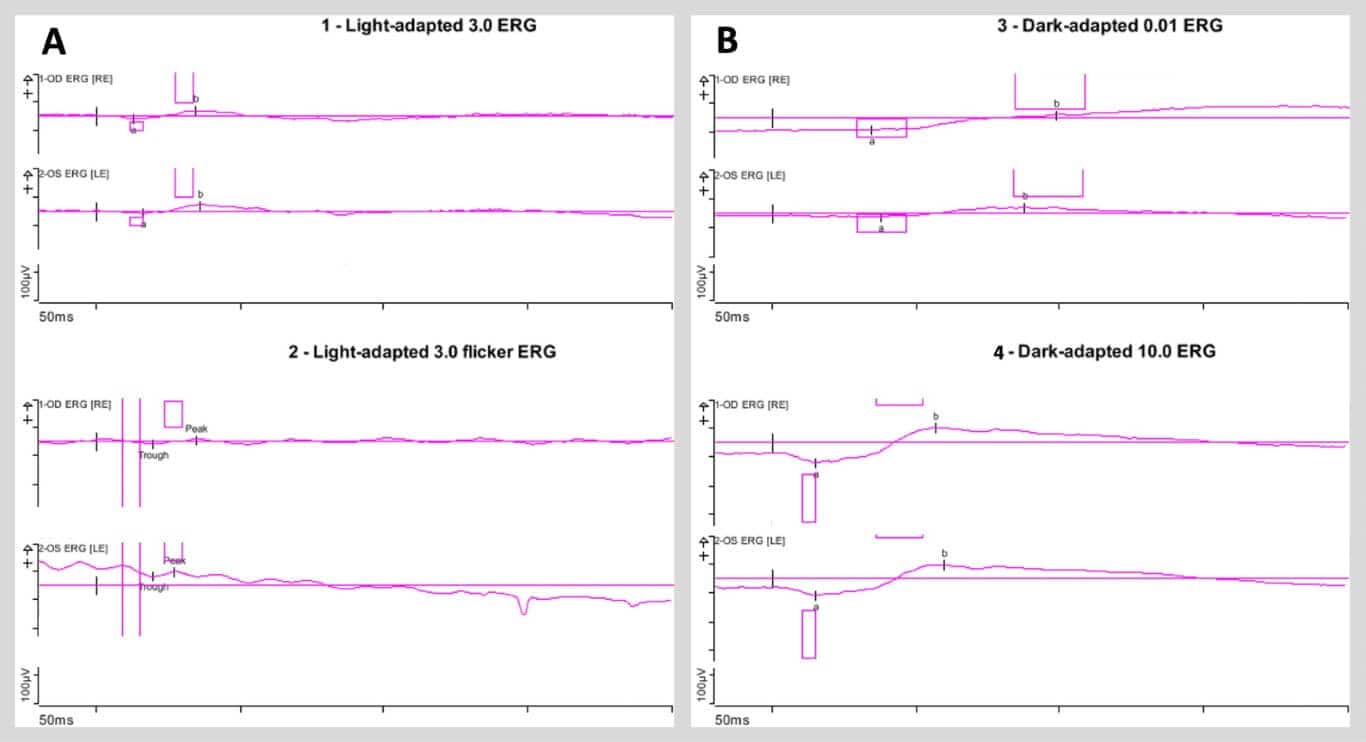
Figure 5. A) Light-adapted full-field flash electroretinogram (ERG) tracings (left panel) of the right and left eyes (top and bottom traces, respectively) demonstrating attenuated amplitudes and delayed kinetics of both the a- and b-waves. Light-adapted full-field flicker ERG tracings (right panel) of the right and left eyes (top and bottom traces, respectively) demonstrating impaired response amplitude but preserved frequency of response in each eye. B) Dark-adapted full-field flash ERG tracings with dim flash (top panel) and bright flash (bottom panel) of the right and left eyes (top and bottom traces in each panel, respectively), both demonstrating attenuated response amplitudes and delayed kinetics of the a- and b-waves in each eye.
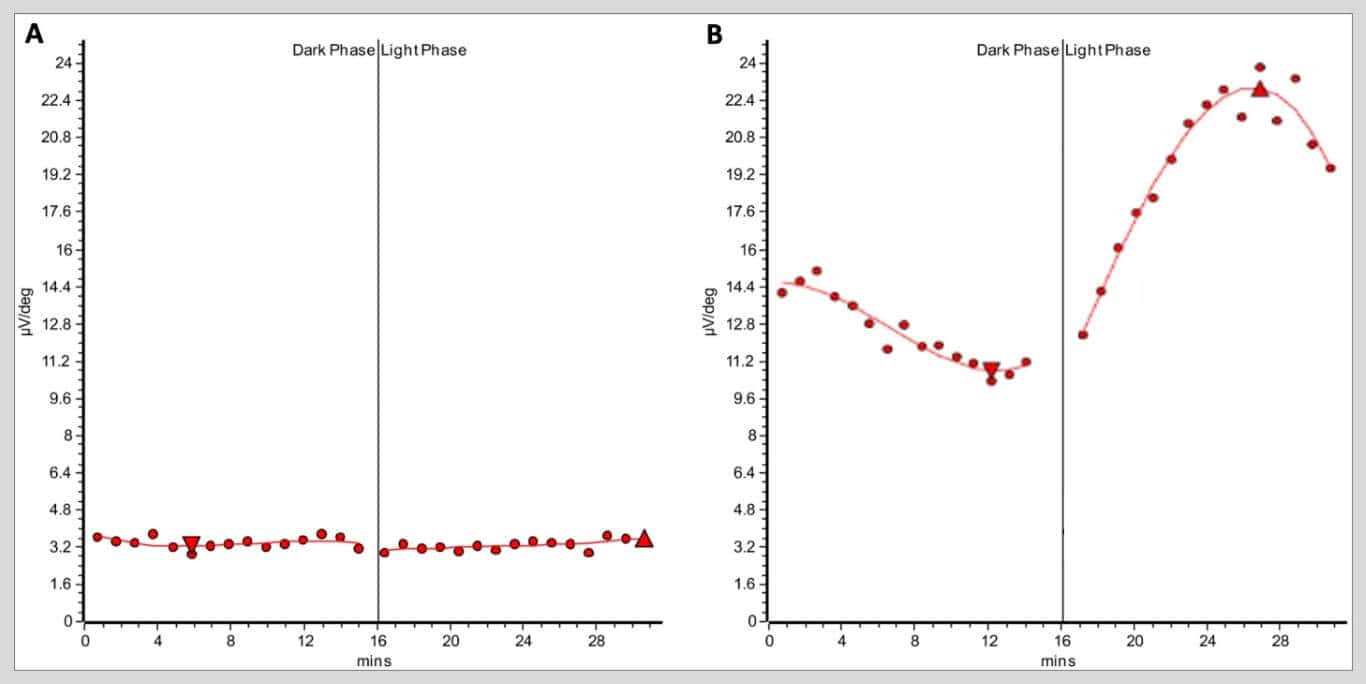
Figure 6. A) Electro-oculogram (EOG) recording of the patient’s right eye demonstrating severely decreased responses with subsequent attenuated light peak-to-dark trough (Arden) ratio. The patient’s EOGs were virtually identical between eyes; the Arden ratio of the right and left eyes were 1.10 and 1.11, respectively. B) A representative normal EOG from a different healthy patient demonstrating a robust response to both dark and light phases with a subsequent normal Arden ratio of 2.2. Normal Arden ratio: >1.80; subnormal: 1.60 – 1.80; reduced: < 1.60.
The patient’s DNA was collected in-office via buccal swab for genetic sequencing for further investigation, given the above findings. Genetic testing revealed positive heterozygous pathogenic variants in both the CTNNA1 gene (c.965C>T (p.Ser322Leu)) and BEST1 gene (c.288G>C (p.Gln96His)). Notably, no mutations in PRPH2 were identified.
Ultimately, a diagnosis of butterfly pattern dystrophy was made based on the characteristic macular appearance, attenuated EOG and ERG responses, and presence of pathogenic CTNNA1 mutation. Although the patient was heterozygous for a known pathogenic BEST1 variant, he did not demonstrate clinical retinal findings (i.e., subfoveal vitelliform lesions)1-3 at the time of examination.This may reflect significantly diminished expressivity or incomplete penetrance of this gene. Given the absence of any macular choroidal neovascularization, hemorrhaging or vitelliform lesions and excellent visual acuity, no intervention was indicated. The patient was instructed to follow up in six months for further retinal evaluation, including repeat fundus photography, fundus autofluorescence, macular OCT, and OCT angiography.
DISCUSSION
First described by Deutman et al. in 1970, BPD, also called butterfly-shaped pattern dystrophy, butterfly-shaped epithelial dystrophy or butterfly-shaped dystrophy of Deutman, is an uncommon form of autosomal-dominant inherited macular pattern dystrophy with unknown prevalence.4,5,9 Most patients retain excellent visual acuity (20/20 – 20/30) throughout life and remain entirely asymptomatic.10 As such, BPD is often identified later in life, usually in the 4th – 5th decade of life.10 Clinically, BPD appears as bilateral symmetric linear, radiating, yellowish macular pigment disruption resembling the wings of a butterfly.9,10 BPD is one of five characterized pattern dystrophies of the macula, the other four including adult-onset foveomacular vitelliform dystrophy (AOFVD), reticular pattern dystrophy, multifocal pattern dystrophy and fundus pulverulentus.4,5 These pattern dystrophies are characterized by various stereotyped patterns of lipofuscin deposition within the RPE, leading to RPE cell dysfunction.4,5
The prototypical gene implicated in all cases of macular pattern dystrophy is PRPH2 (peripherin-2), also known as RDS (retinal degeneration slow).4 Despite sharing a common genetic basis, the pattern dystrophies often present with heterogenous phenotypes that may progress over time and, although uncommon, may lead to vision loss later in life secondary to macular atrophy or choroidal neovascularization.4,6 However, most patients retain good vision throughout their lives without complication.6
In 2016, Saksens et al.7 revealed a novel genetic cause of BPD in CTNNA1, a gene which encodes α-catenin 1. This protein is involved in forming inter-epithelial cell structural adherens junctions and is an important structural component of inter-RPE cell adhesion.7 Although the current literature is sparse regarding reports of the natural history and clinical outcomes of CTNNA1-associated BPD, it appears that most patients retain good vision without complication. However, cases of central vision loss secondary to foveal atrophy11 and choroidal neovascular membrane (CNVM) development12 have been reported. Notably, PRPH2 mutation(s) were not identified in this patient or either of his children, implying that the underlying genetic cause of his BPD can solely be attributed to the mutated CTNNA-1.
In contrast, BVMD is an autosomal dominant inherited macular dystrophy usually presenting early in life, with a prevalence of approximately 1 in 10,000 individuals in the United States.1,2,3,13 BVMD is caused by mutations in the BEST1 gene located on chromosome 11 (11q12.3).1,2,3,13 BEST1 encodes the protein bestrophin-1, a calcium-sensitive chloride channel located on the basolateral aspect of the RPE responsible for maintaining the fluid gradient between the avascular outer neurosensory retina and highly vascular choriocapillaris.1,2,3,13
BVMD classically exhibits a well-defined progressive clinical course with initial “egg yolk” vitelliform lesion development, subsequent collapsing of this lesion and eventual subfoveal outer retinal and RPE atrophy leading to central vision loss.1,2,3,13 Additionally, there is a high risk of CNVM development throughout the clinical course of the disease, with a recent study reporting up to 50% of patients harboring a CNVM as detected on OCT angiography.14 As such, vision loss in BVMD is common.3,13
Both BVMD and BPD are classically inherited in an autosomal dominant manner. However, with autosomal dominant inheritance, 100% penetrance (that is, a given genotype will always produce a given phenotype) and 100% expressivity (that is, the severity of a phenotype produced by a given genotype) cannot always be assumed. Decreased expressivity of a dominant trait may result in a milder phenotype. It has been previously demonstrated that BEST1 mutations causing BVMD exhibit highly variable penetrance and expressivity depending on the exact pathogenic mutation present.15 In fact, over 300 different pathogenic mutations in BEST1 have been described to date.16 It is then possible, and rather likely, that the pathogenic mutations in the BEST1 and CTNNA1 genes of the patients in this case exhibited low penetrance, accounting for the discordant phenotypes between the son, father and daughter. That is, greater penetrance and expressivity of BEST1 in the patient’s daughter, who developed severe BVMD, and very low or even absent penetrance in the patient himself (the father), who has not developed a BVMD phenotype.
Interestingly, a report in 1982 by Gutman et al. described a single case of a young woman who exhibited BPD in one eye and BVMD in the fellow eye simultaneously. Unfortunately, genetic testing was not available at this time, and the family ocular history of the patient was unknown. A subsequent paper (Giuffre and Lodato, 1986) described three siblings who also exhibited simultaneous yet distinct BVMD and BPD phenotypes; in fact, two of the three siblings also showed a BPD-like dystrophy in one eye and a BVMD-like vitelliform macular lesion in the fellow eye. Again, genetic testing was not available at this time for these patients.
BPD has been previously reported to show abnormal EOG recordings (reduced Arden ratio), a measure of RPE health and function.8,17,18,19 Similarly, this patient demonstrated a markedly reduced Arden ratio on EOG testing (see Figure 5), corroborating these previous reports. Interestingly, the patient’s son, whose fundus appeared completely unaffected in each eye on multimodal retinal imaging, exhibited borderline abnormal Arden ratios in each eye. The son shared the same pathogenic mutation in CTNNA1 as his father; however, he notably did not harbor a mutated copy of BEST1. Thus, it appears an impaired EOG is a consistent finding in individuals harboring pathogenic CTNNA1 mutations with or without fulminant BPD.
Further electrophysiologic testing of this patient with full-field ERG revealed attenuated photopic and scotopic a- and b-wave amplitudes (see Figure 5). Hannan et al. (2013)20 previously reported abnormal pattern ERG recordings in 84% of eyes with macular pattern dystrophy, including those with BPD. These authors suggested that such electroretinographic changes may implicate retinal dysfunction beyond RPE dysfunction in macular pattern dystrophy, including BPD. In a mouse model of CTNNA1-associated BPD, researchers found evidence of disrupted ellipsoid zone and external limiting membrane (ELM) in the outer retina, indicative of photoreceptor loss.7 Thus, though generally classified as an isolated dystrophy of the RPE, BPD may affect photoreceptors in the neurosensory retina measurable using standard clinical ERG testing.
Ultimately, commercially available genetic testing with next-generation sequencing was performed to obtain definitive genotypic data for the patients in this case. The genetic data aided in the diagnosis of BPD, better informed its visual prognosis, and helped provide the recognition of potential complications, chiefly vision loss secondary to the development of CNVM and/or macular atrophy.6,11 Moreover, obtaining genetic data for both of the patient’s offspring, particularly in the unaffected son, guided the management plan for close monitoring of the aforementioned complications.
CONCLUSION
BPD and BVMD are distinct entities with disparate genetic underpinnings and phenotypic presentations. However, these macular dystrophies may share a similar underlying pathophysiology as evidenced by previous reports of shared phenotypes between the eyes of the same individual. To these writers’ knowledge, this is the first published report describing a pedigree with identical pathogenic mutations in both CTNNA1 and BEST1 but expressing distinct and prototypical BVMD and BPD phenotypes between individuals. Because both conditions are dominantly inherited, it is uncommon for such unrelated phenotypes to present from simultaneous dominant genotypes. Such genotype-phenotype discordance may be explained by decreased genetic penetrance and/or expressivity. As such, this case emphasizes the importance of genetic testing in cases of suspected inherited retinal disease for accurate diagnosis and prognostication of disease. Abnormal EOG and ERG were demonstrated as findings in BPD; this case also highlights the risk for development of choroidal neovascularization and subsequent risk of vision loss in patients with BPD.
REFERENCES
- Bianco L, Arrigo A, Antropoli A, Berni A, Saladino A, Vilela MA, et al. Multimodal imaging in Best vitelliform macular dystrophy: Literature review and novel insights. Eur J Ophthalmol. 2024 Jan;34(1):39-51.
- Boon CJ, Theelen T, Hoefsloot EH, van Schooneveld MJ, Keunen JE, Cremers FP, et al. Clinical and molecular genetic analysis of Best vitelliform macular dystrophy. Retina. 2009 Jun;29(6):835-47.
- Laich Y, Georgiou M, Fujinami K, Daich Varela M, Fujinami-Yokokawa Y, Hashem SA, et al. Best vitelliform macular dystrophy natural history study report 1: Clinical features and genetic findings. Ophthalmology. 2024 Jul;131(7):845-54.
- Francis PJ, Schultz DW, Gregory AM, Schain MB, Barra R, Majewski J, et al. Genetic and phenotypic heterogeneity in pattern dystrophy. Br J Ophthalmol. 2005 Sep;89(9):1115-9.
- Hanif AM, Yan J, Jain N. Pattern dystrophy: An imprecise diagnosis in the age of precision medicine. Int Ophthalmol Clin. 2019;59(1):173-94.
- Zhang K, Garibaldi DC, Li Y, Green WR, Zack DJ. Butterfly-shaped pattern dystrophy: A genetic, clinical, and histopathological report. Arch Ophthalmol. 2002 Apr;120(4):485-90.
- Saksens NT, Krebs MP, Schoenmaker-Koller FE, Hicks W, Yu M, Shi L, et al. Mutations in CTNNA1 cause butterfly-shaped pigment dystrophy and perturbed retinal pigment epithelium integrity. Nat Genet. 2016 Feb;48(2):144-51.
- Robson AG, Frishman LJ, Grigg J, Hamilton R, Jeffrey BG, Kondo M, Li S, McCulloch DL. ISCEV Standard for full-field clinical electroretinography (2022 update). Doc Ophthalmol. 2022 Jun;144(3):165-177.
- Deutman AF, van Blommestein JD, Henkes HE, Waardenburg PJ, Solleveld-van Driest E. Butterfly-shaped pigment dystrophy of the fovea. Arch Ophthalmol. 1970 May;83(5):558-69.
- Kumar V, Kumawat D. Multimodal imaging in a case of butterfly pattern dystrophy of retinal pigment epithelium. Int Ophthalmol. 2018 Apr;38(2):775-9.
- Tanner A, Chan HW, Pulido JS, Arno G, Ba-Abbad R, Jurkute N, et al. Clinical and genetic findings in CTNNA1-associated macular pattern dystrophy. Ophthalmology. 2021 Jun;128(6):952-5.
- Świerczyńska M, Danikiewicz-Zagała M, Sedlak L, Nowak-Wąs M, Wyględowska-Promieńska D. Choroidal neovascularization associated with butterfly-shaped pattern dystrophy – a case report. Rom J Ophthalmol. 2023;67(2):185-90.
- Laich Y, Georgiou M, Fujinami K, Varela MD, Fujinami-Yokokawa Y, Hashem SA, et al. Best vitelliform macular dystrophy natural history study report 2: Fundus autofluorescence and optical coherence tomography. Ophthalmol Retina. 2025 Mar 12:S2468-6530(25):103-4.
- Han IC, Coussa RG, Mansoor M, Critser DB, Sohn EH, Russell JF, et al. Choroidal neovascularization is common in Best vitelliform macular dystrophy and plays a role in vitelliform lesion evolution. Ophthalmol Retina. 2023 May;7(5):441-9.
- Boon CJ, Klevering BJ, Leroy BP, Hoyng CB, Keunen JE, den Hollander AI. The spectrum of ocular phenotypes caused by mutations in the BEST1 gene. Prog Retin Eye Res. 2009 May;28(3):187-205.
- Nachtigal AL, Milenkovic A, Brandl C, Schulz HL, Duerr LMJ, Lang GE. Mutation-dependent pathomechanisms determine the phenotype in the bestrophinopathies. Int J Mol Sci 2020;21(5):1597.
- Gutman I, Walsh JB, Henkind P. Vitelliform macular dystrophy and butterfly-shaped epithelial dystrophy: A continuum? Br J Ophthalmol. 1982 Mar;66(3):170-3.
- Giuffrè G, Lodato G. Vitelliform dystrophy and pattern dystrophy of the retinal pigment epithelium: concomitant presence in a family. Br J Ophthalmol. 1986 Jul;70(7):526-32.
- Prensky JG, Bresnick GH. Butterfly-shaped macular dystrophy in four generations. Arch Ophthalmol. 1983 Aug;101(8):1198-203.
- Hannan SR, de Salvo G, Stinghe A, Shawkat F, Lotery AJ. Common spectral domain OCT and electrophysiological findings in different pattern dystrophies. Br J Ophthalmol. 2013 May;97(5):605-10.

Zachary Turple, OD, MSc, FAAO currently serves as a staff optometrist at the Joslin Diabetes Center's Beetham Eye Institute at the Beth Israel Deaconess Medical Center in Boston, MA. A native of Vancouver Island, Canada, Dr. Turple obtained his optometry degree from the New England College of Optometry in 2024 before completing a residency in ocular disease with an emphasis in retina and glaucoma at SUNY College of Optometry in New York City.

Dr. Bass is a Distinguished Teaching Professor and a 45-year member of the faculty at the SUNY College of Optometry. She is an attending in the Retina and Electrodiagnostic Clinic and Residency Supervisor of the SUNY Residency in Ocular Disease.


{kind=link}