




A Case of Multiple Evanescent White Dot Syndrome in a Myopic Male
 Save as PDF
Save as PDFABSTRACT
BACKGROUND
Multiple evanescent white dot syndrome (MEWDS) is a rare, often unilateral, primary inflammation of the choriocapillaris, with a predilection for young, healthy females. While self-limiting, MEWDS usually presents with symptoms of central visual disturbances and/or loss, which can be associated with a myriad of diagnoses. Therefore, multimodal imaging studies are necessary to confirm the diagnosis of MEWDS. As imaging techniques continually evolve, optometrists have gained a better understanding of MEWDS pathophysiology and its categorization into primary and secondary forms.
CASE REPORT
A 35-year-old male with a history of pathologic myopia presented for examination two days after experiencing a sudden, painless, paracentral vision loss in his right eye. The ocular examination identified an abnormal Amsler grid defect in the right eye. The macular optical coherence tomography (OCT) study revealed disorganization of the outer retinal layers in the central subfield. His one-week follow-up exam included fundus autofluorescence (FAF) testing, which detected focal areas of hyperautofluorescence surrounding the macula and extending from the posterior pole to the mid-periphery. Approximately 4-6 weeks after the initial onset of symptoms, visual symptoms and improved and serial imaging showed resolution of the fundus pathology.
CONCLUSION
This paper details a case of MEWDS in a seldom-affected gender and emphasizes the benefit of early, multimodal diagnostic imaging to confirm the diagnosis and monitor disease progression. Additionally, this case details the categorization of secondary, or Epi-MEWDS, which can be associated with pathologic myopic findings.
Keywords: MEWDS, Epi-MEWDS, secondary MEWDS, multimodal imaging, fundus autofluorescence, white dot syndrome, pathologic myopia
INTRODUCTION
Multiple evanescent white dot syndrome (MEWDS) is an acute, self-limiting, non-infectious inflammatory condition. Oftentimes unilateral, MEWDS commonly presents with blurred vision, paracentral or central vision loss, photopsia, and/or floaters.1 MEWDS presents in young adults and has an incidence rate of 0.22 per 100,000 annually with a high predilection for females (5:1).1,2,3 Studies indicate that approximately thirty to fifty percent of patients affected by the condition experience a viral prodrome.1 Ocular findings associated with MEWDS include multifocal, ill-defined, gray-white or yellow-white retinal lesions located in the outer retina.2,4 The lesions are typically found surrounding the fovea and extending into the posterior pole.1,3,5 Additional findings associated with MEWDS may include: foveal granularity, a low-grade vitritis, visual acuity ranging from 20/20 to 20/400, and a relative afferent pupillary defect.1,5 The prognosis is good, with resolution of visual symptoms without treatment within weeks to months, and infrequent recurrences.1,5,6 Newer pathophysiological categorizations divide MEWDS into primary and secondary forms.7,8 While secondary MEWDS often presents with similar characteristics to the primary form, it can occur alongside other chorioretinal pathologies.7,8
CASE REPORT
A 35-year-old white male presented with complaints of painless “missing and shimmering” vision temporal to central fixation in the right eye, which had worsened over three days. The patient described his symptom as an “after-image of a camera flash.” His medical history included anxiety and attention-deficit/hyperactivity disorder. He denied a recent history of systemic infections or viral illness and was immunized for influenza nine months before his reported visual symptoms. His ocular history was remarkable for a congenital red-green color deficiency and high myopia, with a refractive error of -8.50-0.25×055 OD and -9.50-0.25×120 OS, prior to his bilateral implantable collamer lens (ICL) surgery four years earlier. The patient remained an axial myope with axial lengths of 27.2 mm OD 27.42 mm OS. His prescribed medications for ADHD and anxiety included escitalopram, extended-release guanfacine, and bupropion hydrochloride.
At initial clinic presentation, his uncorrected distance and near visual acuities were 20/20 OD and 20/20 OS. Amsler grid test revealed paracentral relative scotomas in each eye. Ocular motility, confrontation fields, and pupil evaluation were unremarkable. The patient’s intraocular pressure (IOP) was 12 mmHg OD and 13 mmHg OS. The biomicroscopy exam showed healthy anterior segment structures, patent laser peripheral iridotomies OU, and clear, centered lens implants OU.
The posterior fundoscopic findings were consistent with previously documented high myopia, including a posterior staphyloma OU. His optic nerves were small with ill-defined, indistinguishable C/D ratios OU. Peripapillary atrophy and focal hyperpigmentation nasal to the optic nerve were apparent in both eyes. Both maculae appeared flat with no apparent lesions or elevation. The peripheral retinal findings were unremarkable with no holes, breaks, or tears OU.
Given that there were no diagnostic findings on fundoscopy, additional testing was obtained, including macular SD-OCT and ultrawide color fundus photographs. The diagnosis was inconclusive, and the patient was instructed to monitor a home Amsler grid and return in one month for repeat diagnostic testing, including fundus autofluorescence (FAF).
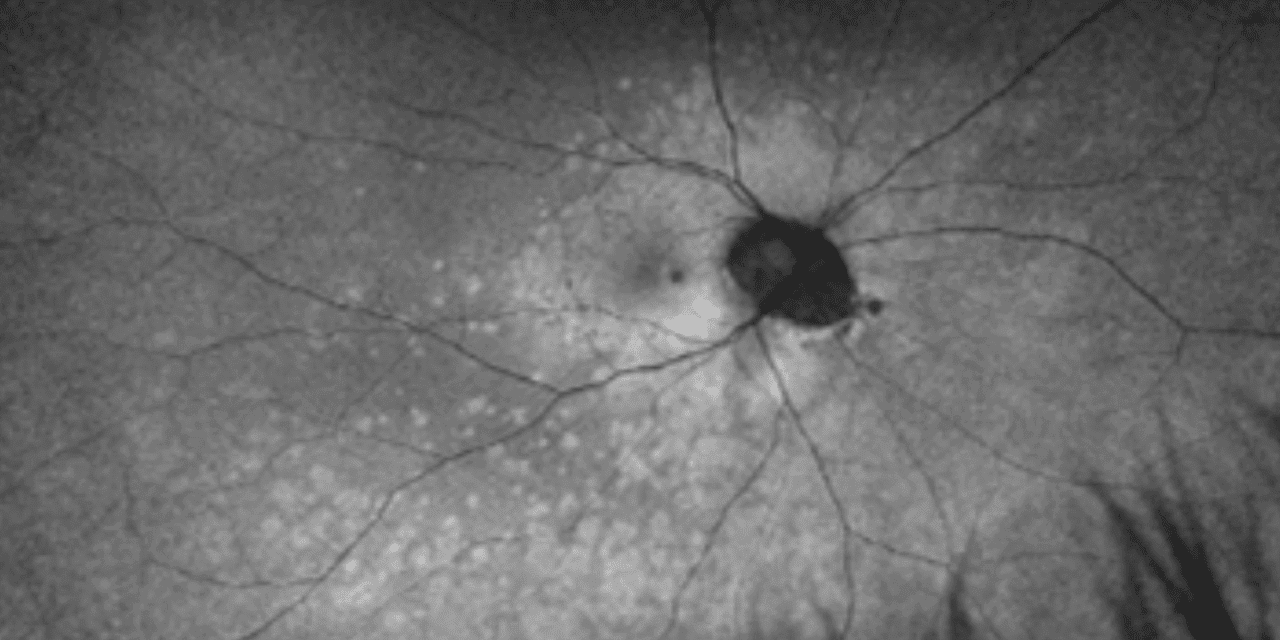
The patient presented one week later with worsening paracentral vision loss, which he described as “constant but inconsistent” scintillating vision loss expanding and contracting throughout the day. He also reported a new onset of photopsia. The patient’s visual acuity remained 20/20 in each eye. The Amsler grid findings showed an increased relative scotoma surrounding central fixation compared to the previous scattered findings. Although the fundus appearance appeared relatively stable (Figures 1A and 1B), the FAF showed multiple dense, pinpoint, hyperfluorescent lesions surrounding the macula and extending into the posterior pole and midperiphery (Figures 1C and 1D). The macular SD-OCT images revealed increased disruption of the deep retinal and retinal pigment epithelium layers of the outer retinal layers, affecting the ellipsoid zone photoreceptors temporal to the foveal depression. Based on the SD-OCT outer retinal pathology and pathognomonic FAF appearance, the patient was diagnosed with MEWDS in his right eye.
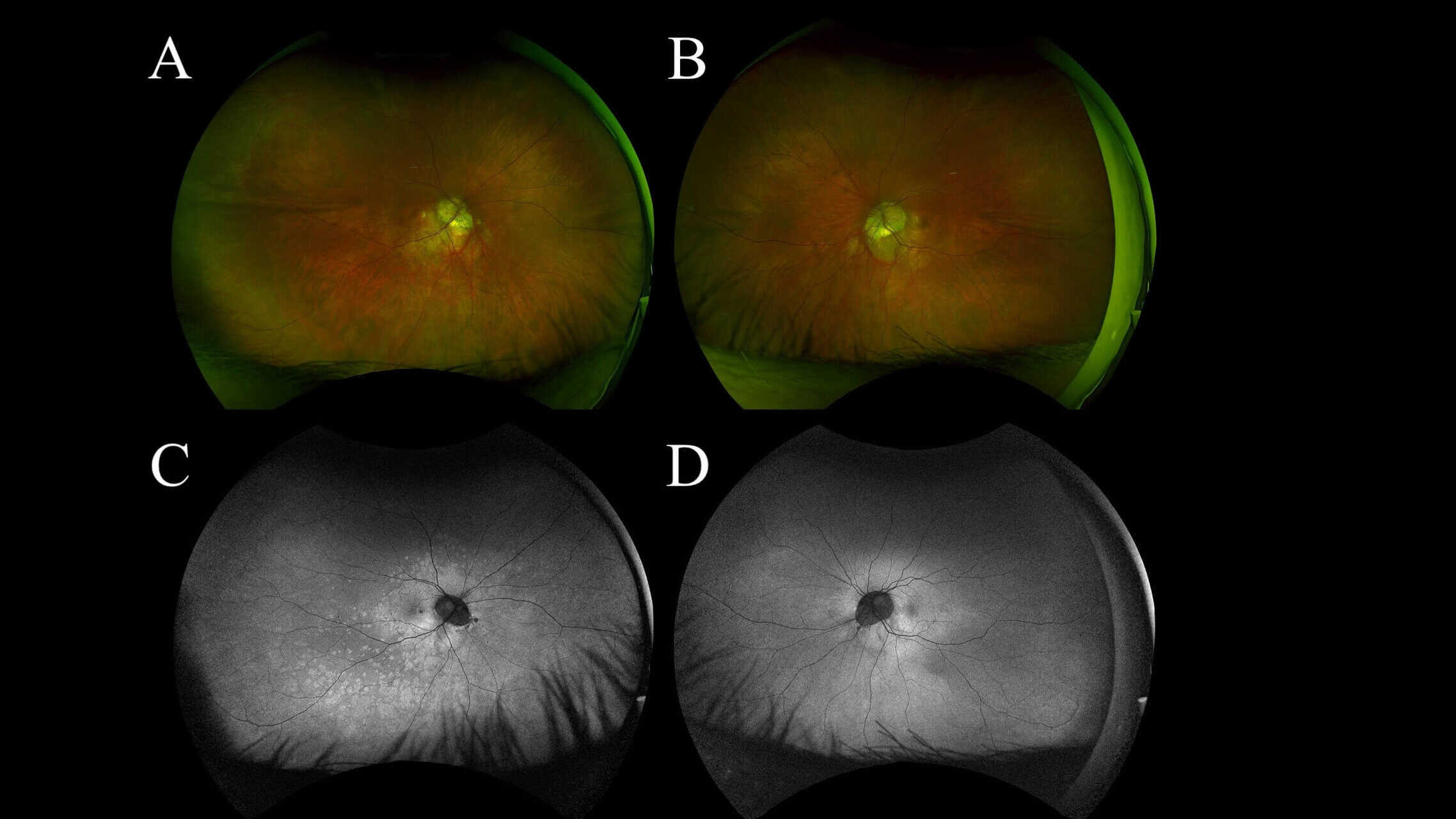
Figure 1: UWF color photo of the right eye (A) and the left eye (B) shows no significant appearance of white lesions at 1 week follow up. WF FAF of the right eye (C) shows small hyperautofluorescent spots and dots in the paramacular area extending through the posterior pole into the midperiphery, no such lesions noted in the left eye (D)
The patient was followed for subsequent one- and three-month follow-ups without indication for treatment. Over the course of the disease process, the macula SD-OCT findings showed improved outer retinal disruption and deposits compared to previous scans. In addition, the FAF results showed progressive parafoveal and posterior pole hypo- and hyperfluorescent changes, with decreased hyperfluorescent intensity. The final three-month follow-up revealed nearly resolved symptoms with infrequent intermittent paracentral photopsia, and continued improvement in the appearance of the diagnostic imaging tests. Figures 2 and 3 summarize the serial testing performed.
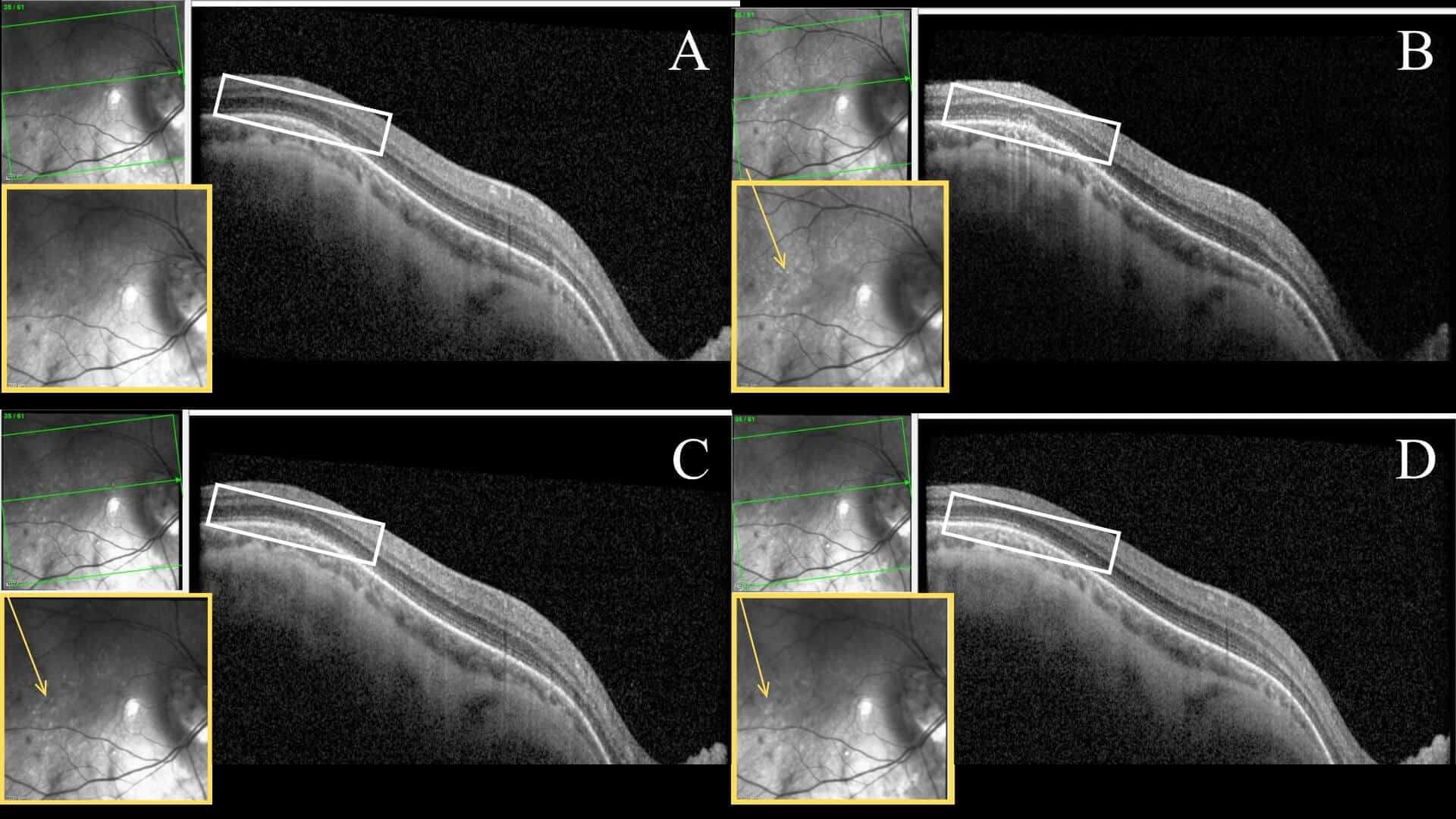
Figure 2: Serial SD-OCT images of the right eye at initial onset (A), one week (B), one month (C), 3 months (D). The white rectangle highlights retinal layers temporal to the fovea that undergo an apparent disruption with an eventual resolution at the EZ layer. The yellow box of each scan shows an enlarged infrared reflectance image. Within this imaging, hyperreflective lesions are visibly scattered throughout the posterior pole, highlighted by the yellow arrow.
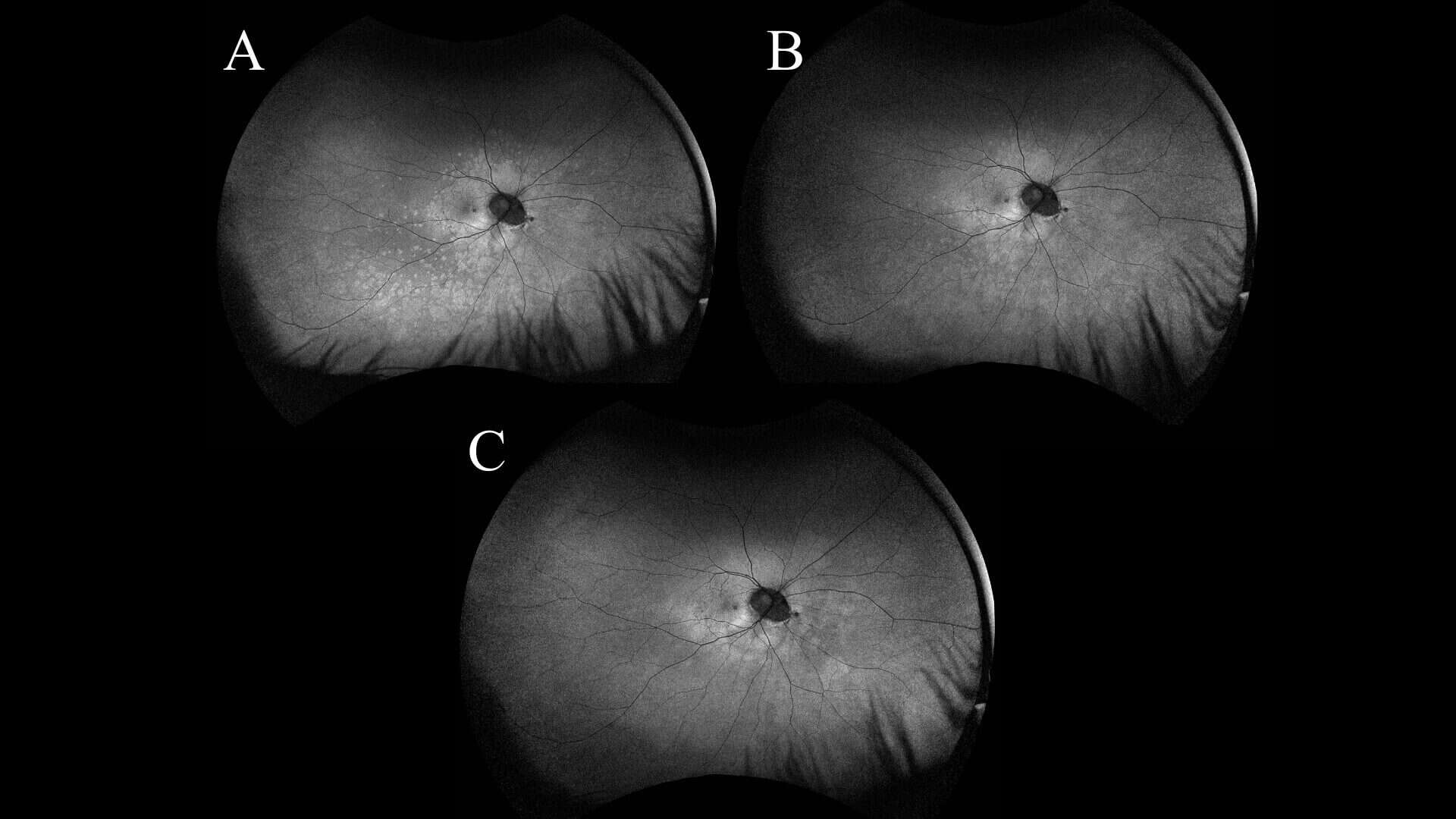
Figure 3: Serial UWF FAF images of the right eye from 1 week (A), 1 month (B), and 3 month follow-ups (C) showing a generalized decrease in hyperautofluorescent lesions with time.
DISCUSSION
Pathophysiology
MEWDS is classified as a primary inflammatory choriocapillaropathy.5 In common with the phenotypical characteristics of other white dot syndromes, the condition presents with scattered multifocal lesions throughout the posterior pole and midperiphery.6 When analyzed, each dot appears to be composed of a cluster of multiple smaller lesions. The white dots tend to align with retinal vessels, leading to the appearance of venous sheathing, and they generally spare the foveal region.9 While historical clinical descriptions of MEWDS place it in a white dot syndrome category, advancements in multimodal imaging have led researchers to determine that MEWDS’ pathophysiology warrants an alternate categorization.2,6 A study by Papasavvas et al. found that Indocyanine green angiography (ICGA) and SD-OCT demonstrate vaso-occlusion at the level of the inner choroid, leading to ischemia in the outer retina and damage to the outer segments of the photoreceptors.10 These findings categorize its mechanism as a choriocapillaritis, as opposed to the stromal chorioretinitis in other white dot syndromes such as birdshot chorioretinitis.5,11,12
The etiology of MEWDS is unknown, but studies suggest it may be immune-mediated.11,13 The inflammation in MEWDS is believed to occur in the peripapillary circulation, causing an enlarged blind spot.8,11 Researchers believe that microglial cells located in the middle-to-inner retina initiate a retinal inflammatory response. Once activated, these microglial cells proliferate and migrate to the site of injury,11 causing morphological changes via signaling and gene expression.11 Recent studies indicate that these microglial cells are responsible for the retinal changes throughout the course of MEWDS.11
Primary vs. Secondary MEWDS
With the advancement in imaging and understanding of MEWDS pathophysiology, two categories of the condition have emerged. A study by Meng et al. described these categories as a primary or idiopathic subtype and a secondary one known as epiphenomenon (Epi-MEWDS).8 This secondary category of the condition is believed to occur as an epiphenomenon of other ocular pathologies from primary MEWDS.8,13 While primary MEWDS is believed to be linked to a viral-like infection, researchers hypothesize that Epi-MEWDS’ pathogenesis is caused by alteration of the outer blood-retina barrier (BRB) resulting in photoreceptor cell exposure to the immune system and an inflammatory response.7,8,13,14,15 The current literature reports the observation of Epi-MEWDS in eyes with a macular neovascular membrane (MNV), leading to the hypothesis that the leaky vessels of MNV may contribute to ongoing outer retinal inflammation.13 In addition, eyes with degenerative myopic findings have been linked to developing Epi-MEWDS due to this inflammatory process.8,13,16
Clinically, Epi-MEWDS demonstrates outer retinal disruption around the presumed triggering retinal lesion and expands centrifugally with an asymmetrical distribution.13 Additionally, multimodal imaging shows similar findings between the two categories, with Epi-MEWDS typically having fewer and smaller lesions.13 Present findings do not show a difference between primary and Epi-MEWDS in gender distribution, visual acuity at presentation, and ocular symptoms.13 However, patients with pre-existing retinal conditions/Epi-MEWDS may not experience the same level of visual recovery as the primary form.13
Diagnostic Imaging
Sources consistently point to multimodal imaging as key to diagnosing either form of MEWDS.4,9,10,13 These sources list SD-OCT, Fluorescein Angiography (FA), Indocyanine Green Angiography (ICGA), FAF, and color fundus photos as the imaging tools for diagnosing MEWDS. There is no consensus on which test or combination is best to confirm a MEWDS diagnosis.5
The appearance of MEWDS on SD-OCT shows lesions at the level of the outer retina; specifically, a disruption of the ellipsoid zone (EZ) and interdigitation zone (IZ) complex in the fovea.3,5,13 Studies also frequently note accumulations of hyperreflective material at the EZ and IZ. These areas form “dome shaped” deposits on top of the RPE. When the lesions resolve spontaneously, residual areas of RPE atrophy are common.5 However, during the acute phase of the disease, RPE and photoreceptors are often spared.17 Additionally, SD-OCT may show an increase and then normalization of choroidal thickness during the acute and recovery phases of MEWDS.5,11
Fluorescein angiography findings associated with MEWDS include early punctate hyperfluorescent lesions that are evident in mid- to late stages. In contrast, hyperfluorescent dots appear in clusters during the choroidal-filling and retinal artery perfusion phases as a “wreath-like” pattern near the macula from the excitement of microglia in immune-mediated inflammation.5,10,11,17 The “wreath-like” pattern is classic for MEWDS, but is not always present, making FA an unreliable tool on its own.5,10
FAF is highly sensitive to the loss of photoreceptor outer segments that occur in MEWDS, which leads to hyperautofluorescent lesions in the MEWDS’ acute phase.1,10,17 This FAF hyperautofluorescence is due to photoreceptor pigment loss, which enhances the underlying RPE autofluorescence.10 FAF plays a significant role in MEWDS diagnosis as a fast, non-invasive imaging modality that co-localizes diseased areas with those found on ICGA.
Color fundus photos are not diagnostic of MEWDS, but they are a tool for documenting the changes in the retinal appearance over the course of the condition.3,10 If visible in photos, gray-white lesions appear at the level of the outer retina and RPE and are scattered through the posterior pole and mid-periphery.5 During the condition’s late stage, foveal granularity may become evident.12
Optical Coherence Angiography (OCTA) is a non-invasive imaging technique that produces high-resolution images of retinal vascular layer blood flow.6 OCTA can be helpful when differentiating MEWDS from white dot syndromes that are associated with MNV. For primary MEWDS, an OCTA should show no choriocapillary morphology.6 In secondary MEWDS, an OCTA could be helpful in confirming a prior retinal inflammatory insult if the imaging had been performed during a previously active MNV.
Case Analysis
With the presence of pathologic myopia and evidence of pre-existing choroidal and retinal disruption, this patient’s MEWDS was categorized as the secondary form, Epi-MEWDS. This patient did not have a viral-prodrome frequently associated with primary MEWDS. Instead, the clinical picture, including the patient’s progressive myopic pathology, was assessed as the etiology of an autoimmune response observed in Epi-MEWDS.8,13,14 FAF imaging detected the classic MEWDS pinpoint hyperautofluorescent macular lesions that were not evident with ophthalmoscopy and color fundus photos. The patient’s history, symptoms, FAF, and SD-OCT findings, collectively confirmed the diagnosis and thus it was deemed not necessary to refer him for invasive fluorescein angiography.
CONCLUSION
This case involving a myopic male with MEWDS supports the contention that multimodal imaging, including FAF and SD-OCT, is essential in making MEWDS diagnoses such as this uncommon presentation. Though self-limiting in nature, MEWDS can easily be misdiagnosed if the condition is not evaluated soon after symptom onset and frequently through the disease process. Optometrists have access to an arsenal of diagnostic tests that help detect conditions not frequently encountered in practice, and FAF and SD-OCT should be considered essential tools when encountering unclear cases of central visual disturbances.
REFERENCES
- Raven ML, Ringeisen AL, Yonekawa Y, Stem MS, Faia LJ, Gottlieb JL. Multi-modal imaging and anatomic classification of the white dot syndromes. Int J Retina Vitr. 2017;3(1):12. doi:10.1186/s40942-017-0069-8
- Classification criteria for multiple evanescent white dot syndrome. Am J Ophthalmol. 2021;228:198-204. doi:10.1016/j.ajo.2021.03.050
- Lewandowski C, Bastian D. MEWDS: a Teaching Case Report | The Journal of Optometric Education. Optom Educ. 2020;45(3). Accessed October 12, 2025. https://journal.opted.org/article/mewds-a-teaching-case-report/
- Munk MR, Stillenmunkes R, Tillmann A, et al. Evidence and Consensus Based Imaging Guidelines in Multiple Evanescent White Dot Syndrome Multimodal imaging in Uveitis (MUV) Taskforce Report 6. Am J Ophthalmol. 2025;278:191-202. doi:10.1016/j.ajo.2025.06.039
- Papasavvas I, Mantovani A, Tugal-Tutkun I, Herbort CP. Multiple evanescent white dot syndrome (MEWDS): update on practical appraisal, diagnosis and clinicopathology; a review and an alternative comprehensive perspective. J Ophthalmic Inflamm Infect. 2021;11:45. doi:10.1186/s12348-021-00279-7
- Chun-Li Chen YZC, Chun-Li Chen YZC. Clinical features and possible pathogenesis of multiple evanescent white dot syndrome with different retinal diseases and events: a narrative review. Int J Ophthalmol. 2024;17(3):583-595. doi:10.18240/ijo.2024.03.23
- Cicinelli MV, Hassan OM, Gill MK, Goldstein D, Parodi MB, Jampol LM. A Multiple Evanescent White Dot Syndrome-like Reaction to Concurrent Retinal Insults. Ophthalmol Retina. 2021;5(10):1017-1026. doi:10.1016/j.oret.2020.12.007
- Meng Y, Zhang Q, Li L, et al. Primary Multiple Evanescent White Dot Syndrome And Multiple Evanescent White Dot Syndrome Secondary To Multifocal Choroiditis/Punctate Inner Choroidopathy: A Comparative Study. Retina Phila Pa. 2023;43(7):1122-1131. doi:10.1097/IAE.0000000000003776
- Sen A, Rao C, Biswas J. An update of multimodal imaging in white dot syndrome. Oman J Ophthalmol. 2024;17(3):325-333. doi:10.4103/ojo.ojo_116_24
- Yung M, Klufas MA, Sarraf D. Clinical applications of fundus autofluorescence in retinal disease. Int J Retina Vitr. 2016;2:12. doi:10.1186/s40942-016-0035-x
- Tavallali A, Yannuzzi LA. MEWDS, Common Cold of the Retina. J Ophthalmic Vis Res. 2017;12(2):132-134. doi:10.4103/jovr.jovr_241_16
- Fabiani C, Shantha J, Gangaputra S, et al. Is it Time to Adopt a New Nomenclature and Classification for White Dot Syndromes Using Multimodal Imaging Techniques? Report 1 from Multimodal Imaging in Uveitis (MUV) Task Force. Ocul Immunol Inflamm. 2025;33(4):561-569. doi:10.1080/09273948.2024.2423870
- Cicinelli MV, Ramtohul P, Marchese A, et al. Latest advances in white spot syndromes: New findings and interpretations. Prog Retin Eye Res. 2023;97:101207. doi:10.1016/j.preteyeres.2023.101207
- De Simone L, Ferraro V, Bolletta E, et al. Multiple Evanescent White Dot Syndrome as Epiphenomenon of Infectious Chorioretinopathies. Ocul Immunol Inflamm. 2025;33(8):1841-1846. doi:10.1080/09273948.2025.2514987
- Essilfie J, Bacci T, Abdelhakim AH, et al. Are There Two Forms Of Multiple Evanescent White Dot Syndrome? Retina Phila Pa. 2022;42(2):227-235. doi:10.1097/IAE.0000000000003288
- Herbort CP, Papadia M, Neri P. Myopia and Inflammation. J Ophthalmic Vis Res. 2011;6(4):270-283.
- Yeh S, Forooghian F, Wong WT, et al. Fundus Autofluorescence Imaging of the White Dot Syndromes. Arch Ophthalmol. 2010;128(1):46-56. doi:10.1001/archophthalmol.2009.368
- Ong AY, Fordwor K, Charbel Issa P. Centrifugal progression of retinal lesions in the early evolution of multiple evanescent white dot syndrome. Graefes Arch Clin Exp Ophthalmol. 2024;262(3):1001-1003. doi:10.1007/s00417-023-06226-7

Dr. Tyler Kitzman is an assistant professor at Southern College of Optometry in Memphis, TN. He is currently the director of the Institution’s Quality Enhancement Plan (QEP) and is a fellow of the American Academy of Optometry (AAO).

Dr. Mary Theresa Taormina is an attending optometrist at the Memphis VAMC. She holds faculty appointments at multiple optometry schools.

Dr. Halie Cottrill Kitzman is an attending optometrist and Director of the Intermediate Low Vision clinic at the Memphis VAMC. She is a fellow of the American Academy of Optometry (AAO) and is Board Certified by the American Board of Certification in Medical Optometry (ABCMO).


{kind=link}