




A Diagnostic Challenge of Scleromalacia Perforans
 Save as PDF
Save as PDFAbstract
Background
The incidence of scleritis, or inflammation of the sclera, is estimated to be 4 per 100,000 person-years and can be classified into two main categories of anterior segment or posterior segment scleritis, with anterior being the most common. Anterior scleritis can be further broken down into non-necrotizing (diffuse or nodular), necrotizing with inflammation, or necrotizing without inflammation (otherwise known as scleromalacia perforans).1,2 Necrotizing scleritis of anterior or posterior involvement encompasses approximately 10-15% of all scleritis cases and is the most severe and potentially sight-threatening.1 This paper will discuss a unique case of scleromalacia perforans that highlights the diagnostic challenge of differentiating posterior scleritis from scleromalacia perforans and the difficulty in determining infectious versus non-infectious origins.
Case Report
A 74-year-old Caucasian male presented to the eye clinic with complaints of redness of his right eye for six months, followed by vision loss in the same eye for the past two months. He denied any pain or light sensitivity but reported a minor scratching feeling for the previous six months. Abnormal anterior and posterior segment findings of the right eye were 4+ bulbar conjunctival injection 360o, nasal scleromalacia with perforation and uveal prolapse, purulent yellow-white discharge, SPK without infiltrates in the nasal cornea, and a large exudative retinal detachment. B-scan revealed an exudative retinal detachment but no thickening of the scleral wall or any additional signs of posterior scleritis. The patient was diagnosed with scleromalacia perforans and referred to a retina and cornea specialist and a rheumatologist for further care and co-management.
Conclusion
This case demonstrates the potential difficulty in separating anterior from posterior scleritis, along with the importance of uncovering whether an episode of scleritis is infectious or non-infectious in origin.
Keywords: Scleritis, scleromalacia perforans, rheumatoid arthritis
Case Report
A 74-year-old Caucasian male presented to the eye clinic with complaints of redness of his right eye for six months, followed by vision loss in the same eye for the past two months. He denied any pain or light sensitivity but reported a minor scratching feeling for six months. Medical history was significant for high cholesterol, hypertension, and rheumatoid arthritis. He was on the following medications: atorvastatin 80mg daily and prednisone 10mg daily. Visual acuity corrected was hand motion right eye and 20/30 left eye. Preliminary testing was significant for an APD and constricted confrontation fields of the right eye. Abnormal anterior and posterior segment findings were 4+ bulbar conjunctival injection 360o, nasal scleromalacia with perforation and uveal prolapse, purulent yellow-white discharge, SPK without infiltrates in the nasal cornea, and a large exudative retinal detachment. Anterior and posterior evaluation of the left eye was normal, with the exception of 1+ nuclear sclerotic cataracts, which was the cause of the reduced acuity of that eye. Intraocular pressures were 20mmHg OD and 16 mmHg OS. B-scan confirmed an exudative retinal detachment without thickening of the scleral wall or any additional signs of posterior scleritis. B-scan measurements of the posterior sclera were 1.11mm OD and 1.15mm OS.

Figure 1: Image of the anterior segment of the right eye showing nasal perforation and uveal prolapse, significant injection 360° and purulent yellow-white discharge.
The patient was referred to retina and cornea specialists, where the eye was cultured. The patient was then referred to rheumatology for improved immunosuppression. The cornea and conjunctival culture revealed a heavy growth of staphylococcus epidermis and moderate growth of streptococcus pneumoniae, so the patient was promptly started on Cephalexin 500mg TID PO and moxifloxacin gtts QID OD. Once the anterior segment infection had cleared, he underwent ocular surface reconstruction with a scleral patch graft and amniotic membrane transplantation. Medications after surgery included prednisolone 10mg PO qDay, moxifloxacin gtts, prednisolone forte gtts 1%, cyclopentolate gtts, and tobramycin gtts and dexamethasone ophthalmic ointment. At the one-month post-operative visit, visual acuity remained hand-motion in the right eye. The retinal detachment had resolved with a retinal fold present through the macula. No retinal treatment was recommended as visual acuity recovery was unlikely. The patient was then placed on methotrexate 15mg weekly in addition to his 10mg qDay prednisone by rheumatology in an effort to improve systemic control of his rheumatoid arthritis. He has not had any recurrences in the past two years.
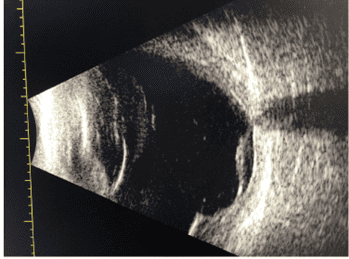
Figure 2: B-Scan ultrasonography of the right eye showing an exudative retinal detachment after scleral graft. There were no B-scan images available prior to the scleral graft.
Discussion
Scleromalacia perforans presents with vague ocular symptoms of irritation without pain. It is most commonly diagnosed in women over the age of 70 with rheumatoid arthritis. Areas of necrosis start near the limbus but can slowly progress to involve thinning of the scleral wall, eventually exposing the uvea.3 Anterior scleritis complications can include stromal keratitis, peripheral ulcerative keratitis, and peripheral corneal thinning.4 In contrast, necrotizing anterior scleritis with inflammation appears similar, but differs in that it often presents with gradual onset of pain that interferes with sleep, radiating to the jaw or brow area.3 In this case, because the patient presented without severe pain with a prior diagnosis of rheumatoid arthritis, he was diagnosed with scleromalacia perforans. Unique to this case is the exudative retinal detachment, suggesting some posterior inflammation. However, there are some discrepancies to be discussed, which made this diagnosis challenging.
Posterior scleritis, which involves inflammation of the posterior sclera, presents with a classic symptom of deep pain that is so severe patients often have difficulty sleeping.5 This pain may or may not worsen with eye movement and may also be accompanied by other signs, such as exudative retinal detachments, choroidal folds, uveal effusion, subretinal mass, optic nerve edema, myositis, elevated intraocular pressure, and proptosis.3,4,5 Approximately 20% of cases present concurrently with anterior scleritis, with diffuse or nodular scleral redness and edema.6 Posterior scleritis can be vision threatening if not treated rapidly, and it is most likely to be associated with systemic disease if diagnosed after age 55, such as rheumatoid arthritis. Ultimately the diagnosis of posterior scleritis is confirmed by B-scan ultrasonography, which demonstrates a thicker scleral wall of at least 2mm with or without nodules or the characteristic T-sign, where the posterior episcleral space is filled with a fluid that extends around the optic nerve.3,5,7 If a B-scan is difficult, an MRI or CT scan can demonstrate scleral thickening while also ruling out other potential causes of orbital inflammation.3 Dong et al., in a retrospective review of 23 posterior scleritis patients who all presented with serous retinal detachments involving the macula, demonstrated how relevant B-scan ultrasonography is to a proper diagnosis of posterior scleritis. In this review, 74% were initially misdiagnosed with other retinal or orbital disorders until a B-scan was performed. The confirmatory B-scan finding was a T-sign, present in all cases.8 Most studies propose a thicker scleral wall of at least 2mm in the involved eye compared to the uninvolved eye or the T-sign to definitively diagnose posterior scleritis.3,5,7 However, Dong et al. and Suhr et al. propose a more novel method of posterior scleritis diagnosis, comparing the scleral thickness between the involved and uninvolved eye. Dong et al.’s data discovered the average difference in scleral thickness between the involved and uninvolved eye to be 2.51 +-0.81mm vs. 1.09+-0.29mm. In contrast, Suhr et al. presented their retrospective review of the same data as 1.94 +- 0.18mm vs 0.94 +- 0.18mm.7,8 These data points suggest that a difference of at least 2mm is not sensitive enough to detect some cases of posterior scleritis, but further research should be performed before changing this diagnostic criteria. With this presented patient, the B-scan did not reveal the T-sign or a thickened scleral wall of 2mm when compared to the non-involved eye, suggesting the patient did not present with posterior scleritis. However, all other causes of exudative retinal detachments, such as other inflammatory, infectious or neoplastic etiologies, were excluded or unlikely through his physical exam and history. In addition, the detachment resolved with anti-inflammatory treatment and improved control of rheumatoid arthritis, further supporting the diagnosis of posterior scleritis. Thus, this case supports Dong et al. and Surh et al. in the need for redefining the confirmatory diagnosis of posterior scleritis.
Scleritis, either anterior or posterior, can arise from various causes, but there are two main categories: infectious and non-infectious. Infectious scleritis comprises approximately 5-10% of cases, whereas 90-95% are of non-infectious origin. Approximately 30-50% of the non-infectious category have an associated autoimmune disease.9 This case is unique in that it had an underlying autoimmune cause and an overlying bacterial infection. It is important as a clinician to be able to distinguish between infectious and non-infectious cases, as treating an infectious case with the typical immunosuppressants can exacerbate the infection and potentially lead to devastating consequences, such as vision loss.
Infectious scleritis can be subdivided into exogenous scleritis, which results from trauma or surgery, and endogenous scleritis, which is less common and a result of systemic infection. Factors that can lead to infection include previous ocular surgery or trauma, chronic use of ophthalmic corticosteroids, systemic immunosuppression, or severe ocular surface diseases. Diagnosis can be challenging without previous ocular surgery or trauma, as infectious scleritis can look comparable to diffuse, nodular, or necrotizing non-infectious scleritis. Clinical signs that point to an infectious etiology include conjunctival ulceration, visible pus or abscesses, purulent exudates, hypopyon, keratic precipitates, or worsening clinical appearance after starting immunosuppressant treatment.9 To aid in the diagnosis of infectious keratitis, scleral or corneal scrapings should be performed only if there is adjacent keratitis or an ulcerated scleral nodule, as unnecessary scrapings could worsen inflammation and delay healing.9
In this case report, the patient had a prior diagnosis of rheumatoid arthritis, making the etiology most likely to be inflammatory. However, due to the mucopurulent discharge and adjacent corneal keratitis, corneal and conjunctival scrapings were performed. Cultures revealed a Staphylococcal overlying infection, so treatment was first started with oral and topical antibiotics.
The two prevalent autoimmune etiologies of scleritis are rheumatoid arthritis (RA) and granulomatosis with polyangiitis (GPA). Scleritis due to RA or GPA can range from mild to severe presentations but is often necrotizing.10 Other known autoimmune diseases that have been reported to cause scleritis include the following: systemic lupus erythematosus, sarcoidosis, Vogt-Koyanagi-Harada disease, inflammatory bowel disease, Takayasu disease, Cogan’s Syndrome, polyarteritis nodosa, vasculitis, Wegener’s, Crohn’s disease, and ulcerative colitis.9,11,12 In this case, the patient has a diagnosis of RA. RA is an autoimmune disorder often characterized by bilateral synovitis and possible extra-articular manifestations, including ocular involvement.13 Scleritis occurs due to the deposition of immune complexes in the scleral and conjunctival space, eventually leading to a local vasculitis and necrotizing scleritis.14 Typically, scleritis does not occur until at least ten years after the initial RA diagnosis and can present bilaterally in approximately 40% of patients. Scleritis is a significant finding for those with RA, as those with scleritis often present with worse joint and extra-articular pathology, thus resulting in higher mortality and morbidity. If not treated systemically, 36-45% of RA patients afflicted with scleritis pass away within three years after the onset of scleritis.4
Treatment of scleromalacia perforans is aimed first at preserving ocular structure and secondarily at controlling the cause of systemic inflammation. The first line of treatment should be a clinical evaluation to determine if there is any infectious cause. In this case, the patient was treated with both oral cephalexin and moxifloxacin 0.5% ophthalmic solution after the clinical signs of keratitis and mucopurulent discharge prompted clinicians to perform corneal scrapings. In this stage, systemic NSAIDS should never be used in necrotizing scleritis for fear of the risk of scleral perforation, and topical steroids only serve to treat any existing edema or pain, which our patient did not experience.15 After any infectious cause is treated, repairing the scleral perforation should be the next goal, as this is necessary to prevent phthisis bulbi.3
While most small scleral weaknesses could resolve with medical therapy alone, larger defects require surgical methods. Typically, patients present later with larger defects due to the lack of or minimal pain upon presentation and thus require ocular surface reconstruction, such as a scleral patch graft and amniotic membrane.10 In addition, it is important to treat with immunosuppressants early to treat the ocular condition as well as systemic inflammation in an attempt to preserve vision.15 Systemic treatments vary widely depending on underlying systemic disease and severity but, in general, include medications within the following classes: systemic steroids, non-biologic disease-modifying antirheumatic drugs, or biologic disease-modifying antirheumatic drugs.15,16 Systemic treatments are often continued once the scleritis is resolved to prevent recurrence and better control the underlying cause of inflammation.16 In this case, the patient was placed on systemic prednisone as well as methotrexate to control his rheumatoid arthritis. It is imperative for the eye care specialist to continue to work closely with the primary care physician or rheumatologist responsible for the patient’s systemic health, as modification to systemic treatment may be necessary if future ocular complications occur.6
Conclusion
Scleritis can be a vision-threatening and life-threatening condition. It is essential that an eye care provider not only recognize this condition but know how to treat and co-manage with specialists, potentially including cornea, retina, primary care, and rheumatology. This case demonstrates the diagnostic challenge of separating anterior from posterior scleritis and how the two etiologies may overlap. When encountering a scleritis case, a practitioner should first determine if the cause is infectious, non-infectious, or a combination of both, as in this case. If infectious, treatment should be directed first toward the infectious agent. If non-infectious, further work-up should be done for an autoimmune component. If a systemic cause is found, treatment should be directed both systemically and ocularly together to achieve the best possible outcome while under the care of eye care providers as well as primary care and rheumatology.
References
- Vergouwen DPC, Rothova A, Berge JCT, et al. Current insights in the pathogenesis of scleritis. Exp Eye Res. 2020;197:108078. doi:10.1016/j.exer.2020.108078
- Cunningham ET Jr, McCluskey P, Pavesio C, et al. Scleritis. Ocul Immunol Inflamm. 2016;24(1):2-5. doi:10.3109/09273948.2016.1136190
- Bowling, B. (2015). Kanski’s clinical ophthalmology (8th ed.). W B Saunders.
- Ghauri MI, Riaz SU, Husain A, et al. Scleromalacia perforans: a case report. J Med Case Rep. 2018;12(1):155. Published 2018 Jun 5. doi:10.1186/s13256-018-1686-z
- Rifkin, L. Posterior Scleritis: A Diagnostic Challenge. Review of Ophthalmology.2018, February 9 https://www.reviewofophthalmology.com/article/posterior-scleritis-a-diagnostic-challenge
- Promelle V, Goeb V, Gueudry J. Rheumatoid Arthritis Associated Episcleritis and Scleritis: An Update on Treatment Perspectives. J Clin Med. 2021;10(10):2118. Published 2021 May 14. doi:10.3390/jcm10102118
- Suhr, K., & Patel, S. (2015). Evaluation of Diagnostic Criteria for B-Scan Ultrasonography in Posterior Scleritis. Investigative Ophthalmology & Visual Science, 56.
- Dong, Z. Z., Gan, Y. F., Zhang, Y. N., Zhang, Y., Li, J., & Zheng, H. H. (2019). The clinical features of posterior scleritis with serous retinal detachment: a retrospective clinical analysis. International journal of ophthalmology, 12(7), 1151–1157. https://doi.org/10.18240/ijo.2019.07.16
- Murthy SI, Sabhapandit S, Balamurugan S, et al. Scleritis: Differentiating infectious from non-infectious entities. Indian J Ophthalmol. 2020;68(9):1818-1828. doi:10.4103/ijo.IJO_2032_20
- Ghafoor SY, Williamson J. Surgical management of scleromalacia perforans–a case report. Scott Med J. 1983;28(4):357-359. doi:10.1177/003693308302800410
- Wu CC, Yu HC, Yen JH, Tsai WC, Liu HW. Rare extra-articular manifestation of rheumatoid arthritis: scleromalacia perforans. Kaohsiung J Med Sci. 2005;21(5):233-235. doi:10.1016/S1607-551X(09)70193-7
- Reddy SC, Tajunisah I, Rohana T. Bilateral scleromalacia perforans and peripheral corneal thinning in Wegener’s granulomatosis. Int J Ophthalmol. 2011;4(4):439-442. doi:10.3980/j.issn.2222-3959.2011.04.22
- American College of Rheumatology Subcommittee on Rheumatoid Arthritis Guidelines. Guidelines for the management of rheumatoid arthritis: 2002 Update. Arthritis Rheum. 2002;46(2):328-346. doi:10.1002/art.10148
- Fong L.P., Sainz de la Maza M., Rice B.A., Kupferman A.E., Foster C.S. Immunopathology of scleritis. Ophthalmology. 1991;98:472–479.
- Yangzes S, Sharma VK, Singh SR, Ram J. Scleromalacia perforans in rheumatoid arthritis. QJM. 2019;112(6):459-460. doi:10.1093/qjmed/hcy295
- Lawuyi LE, Gurbaxani A. Refractory necrotizing scleritis successfully treated with adalimumab. J Ophthalmic Inflamm Infect. 2016;6(1):37. doi:10.1186/s12348-016-0107-y

Danielle Toms, OD graduated from NOVA Southeastern College of Optometry in 2014 and completed her residency at the Kansas City VAMC in ocular disease and low vision rehabilitation. She has been employed at the Kansas City VAMC since she completed residency in 2015, starting at the Warrensburg CBOC before moving to the main hospital as the Clinical Lead of the Visual Impairment Services Program. She currently serves as the Clinical Lead of Tele-eye and TECS at the KCVA and is ABO certified.

Dr. Elizabeth Phillips graduated from UMSL College of Optometry in 2016. The following year she completed her ocular disease and low vision residency at the Kansas City VA Medical Center, where she currently works, teaching students and residents. She is board certified in medical optometry (ABCMO) and is pursuing her fellowship with AAO this fall.



{kind=link}